Поиск схожих последовательностей в базах данных. Локальное выравнивание. Семейство программ серии BLAST
- Поиск схожих последовательностей в базах данных. Локальное выравнивание. Семейство программ серии BLAST

Содержание
- 2. Выравнивание последовательностей —размещение двух или более последовательностей ДНК, РНК или белков друг под другом таким образом,
- 3. Цель выравнивания – добиться максимального количества совпадений. Что лучше? Просто написать последовательности друг под другом Двигать
- 4. Алгоритм локального выравнивания был предложен Т. Ф. Смитом и М. Уотерменом в 1981 г. В обоих
- 5. – Стоимость замены, вcтавки, делеции Значительно чаще 1 длинная делеция, чем много коротких => штраф за
- 6. AACGTTTCCAGTCCAAATAGCTAGGC ===--=== =-===-==-====== AACCGTTC TACAATTACCTAGGC Hits(+1): 18 Misses (-2): 5 Gaps (existence -2, extension -1): 1
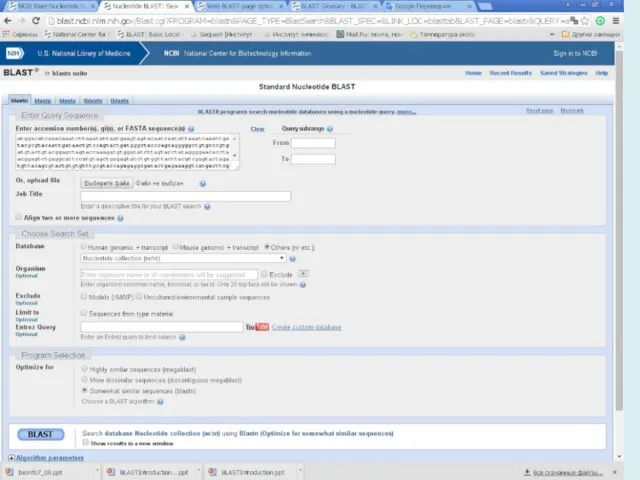
- 7. http://blast.ncbi.nlm.nih.gov/Blast.cgi BLAST (Basic Local Alignment Search Tool) — семейство компьютерных программ, служащих для поиска гомологов белков
- 8. Используя BLAST, можно сравнить имеющуюся последовательность с последовательностями из базы данных и найти последовательности предполагаемых гомологов.
- 9. Принципы работы BLAST 1. Для каждого слова длины W в искомой последовательности составляется список схожих слов,
- 10. 2. Для каждого слова обрабатываем составленный для него список схожих слов - ищем, по заранее построенной
- 11. 3. Расширяем выравнивание вправо и влево от найденных “затравок” используя алгоритм динамического программирования сначала без гэпов
- 13. После максимального продления размеров всех возможных «слов» изучаемой последовательности, определяются выравнивания с максимальным количеством совпадений для
- 18. ДНК: megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для длинных похожих последовательностей. Оптимален для поиска
- 20. Пример последовательности в формате FASTA : >OVAX_CHICK GENE X PROTEIN QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAEKMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVY Пустые строки в середине не
- 21. Вырожденные нуклеотидные коды (красный цвет), рассматриваются как «mismatches» нуклеотидного выравнивания. Для тех программ, которые используют аминокислотные
- 22. Nucleotide Sequence Databases Nr All GenBank + RefSeq Nucleotides + EMBL + DDBJ + PDB sequences
- 23. Peptide Sequence Databases Nr All non-redundant GenBank CDS translations + RefSeq Proteins + PDB + SwissProt
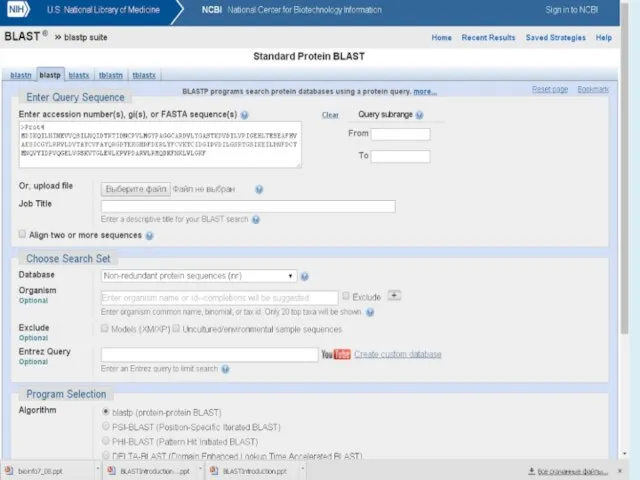
- 25. Белок: PSI-BLAST (Position-Specific Iterated -BLAST) поиск удаленных белковых гомологов с использованием PSSM (position-specific scoring matrix) PHI-BLAST
- 27. При определении сходства ключевым элементом является матрица замен, так как она определяет показатели сходства для любой
- 28. Выбор параметров Меняйте параметры только, если по умолчанию не работает (параметры по умолчанию подобраны хорошо для
- 29. Выбор параметров Матрица:BLOSUM для локального выравнивания обычно лучше, чем PAM Чем выше номер BLOSUM – тем
- 33. E-value (the expectation value) – представляет число различных элайментов с баллами эквивалентными или лучшими, чем S,
- 39. PSI – BLAST Алгоритм: Несколько раундов поиска Первый раунд – просто blastp (BLOSUM62) Построение PSSM на
- 41. DELTA – BLAST (Domain Enhanced Lookup Time Accelerated BLAST)
- 48. MEGAN V5.11.3 http://ab.inf.uni-tuebingen.de/software/megan/ Основное применение программы для разбора и анализа результата BLAST сравнения набора ридов против
- 49. MEGAN V5.11.3 http://ab.inf.uni-tuebingen.de/software/megan/ The aim of MEGAN is to provide a tool for studying the taxonomic
- 50. MEGAN V5.11.3 http://ab.inf.uni-tuebingen.de/software/megan/ However, the main application of the program is to parse and analyze the
- 51. MEGAN V5.11.3 http://ab.inf.uni-tuebingen.de/software/megan/ New input to the program is usually provided as a BLAST file obtained
- 54. DNA Master http://cobamide2.bio.pitt.edu/ DNA Master это Multiple Document Interface (MDI) программа для создания, модификации и анализа
- 55. domain A discrete portion of a protein assumed to fold independently of the rest of the
- 56. RAST (Rapid Annotation using Subsystem Technology) is a fully-automated service for annotating bacterial and archaeal genomes.
- 58. LOCUS FJ795028 701 bp mRNA linear PRI 06-APR-2009 DEFINITION Homo sapiens tumor necrosis factor alpha (TNF)
- 59. CDS /gene="TNF" /note="APC1 protein" /codon_start=3 /product="tumor necrosis factor alpha" /protein_id="ACO37640.1" /db_xref="GI:226201421" /translation="STESMIRDVELAEEALPKKTGGPQGSRRCLFLSLFSFLIVAGAT TLFCLLHFGVIGPQREEFPRDLSLISPLAQAVRSSSRTPSDKPVAHVVANPQAEGQLQ WLNRRANALLANGVELRDNQLVVPSEGLYLIYSQVLFKGQGCPSTHVLLTHTISRIAV SYQTKVNLLSAIKSPCQRETPEGAEAKPWYEPIYLGGVFQLEKGDRLSAEINRPDYLD FAESGQVYFGIIAL«
- 61. Скачать презентацию

Выравнивание последовательностей —размещение двух или более последовательностей ДНК, РНК или белков
Выравнивание последовательностей —размещение двух или более последовательностей ДНК, РНК или белков

Цель выравнивания – добиться максимального количества совпадений. Что лучше?
Просто написать последовательности
Цель выравнивания – добиться максимального количества совпадений. Что лучше?
Просто написать последовательности

Алгоритм локального выравнивания был предложен Т. Ф. Смитом и М. Уотерменом в 1981 г.
В
Алгоритм локального выравнивания был предложен Т. Ф. Смитом и М. Уотерменом в 1981 г.
В

– Стоимость замены, вcтавки, делеции
Значительно чаще 1 длинная делеция, чем много
– Стоимость замены, вcтавки, делеции
Значительно чаще 1 длинная делеция, чем много

AACGTTTCCAGTCCAAATAGCTAGGC
===--=== =-===-==-======
AACCGTTC TACAATTACCTAGGC
Hits(+1): 18
Misses (-2): 5
Gaps (existence -2, extension -1):
===--=== =-===-==-======
AACCGTTC TACAATTACCTAGGC
Hits(+1): 18
Misses (-2): 5
Gaps (existence -2, extension -1):

http://blast.ncbi.nlm.nih.gov/Blast.cgi
BLAST (Basic Local Alignment Search Tool) — семейство компьютерных программ,
http://blast.ncbi.nlm.nih.gov/Blast.cgi
BLAST (Basic Local Alignment Search Tool) — семейство компьютерных программ,

Используя BLAST, можно сравнить имеющуюся последовательность с последовательностями из базы данных
Используя BLAST, можно сравнить имеющуюся последовательность с последовательностями из базы данных

Принципы работы BLAST
1. Для каждого слова длины W в искомой последовательности
Принципы работы BLAST
1. Для каждого слова длины W в искомой последовательности
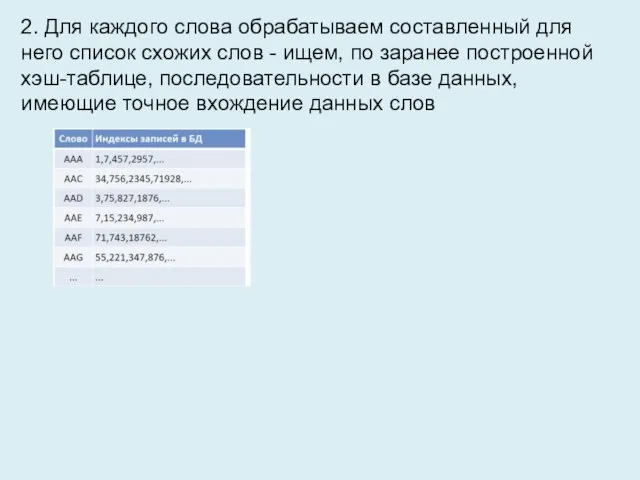
2. Для каждого слова обрабатываем составленный для него список схожих слов
2. Для каждого слова обрабатываем составленный для него список схожих слов

3. Расширяем выравнивание вправо и влево от найденных “затравок” используя алгоритм
3. Расширяем выравнивание вправо и влево от найденных “затравок” используя алгоритм


После максимального продления размеров всех возможных «слов» изучаемой последовательности, определяются выравнивания
После максимального продления размеров всех возможных «слов» изучаемой последовательности, определяются выравнивания

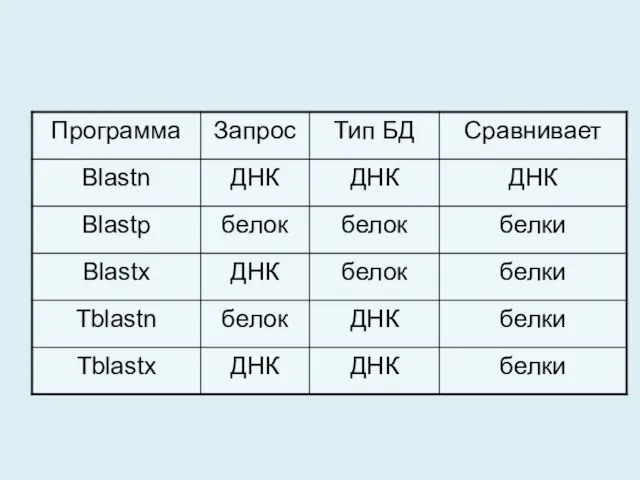


ДНК:
megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для длинных
ДНК:
megaBLAST – другой алгоритм для сравнения ДНК. Оптимизирован для длинных

Пример последовательности в формате FASTA :
>OVAX_CHICK GENE X PROTEIN QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAEKMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVY
Пустые строки
Пример последовательности в формате FASTA :
>OVAX_CHICK GENE X PROTEIN QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAEKMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVY
Пустые строки

Вырожденные нуклеотидные коды (красный цвет), рассматриваются как «mismatches» нуклеотидного выравнивания.
Для тех
Вырожденные нуклеотидные коды (красный цвет), рассматриваются как «mismatches» нуклеотидного выравнивания.
Для тех

Nucleotide Sequence Databases
Nr All GenBank + RefSeq Nucleotides + EMBL +
Nucleotide Sequence Databases
Nr All GenBank + RefSeq Nucleotides + EMBL +

Peptide Sequence Databases
Nr All non-redundant GenBank CDS translations + RefSeq Proteins
Peptide Sequence Databases
Nr All non-redundant GenBank CDS translations + RefSeq Proteins


Белок:
PSI-BLAST (Position-Specific Iterated -BLAST) поиск удаленных белковых гомологов с использованием PSSM
PSI-BLAST (Position-Specific Iterated -BLAST) поиск удаленных белковых гомологов с использованием PSSM


При определении сходства ключевым элементом является матрица замен, так как она
При определении сходства ключевым элементом является матрица замен, так как она

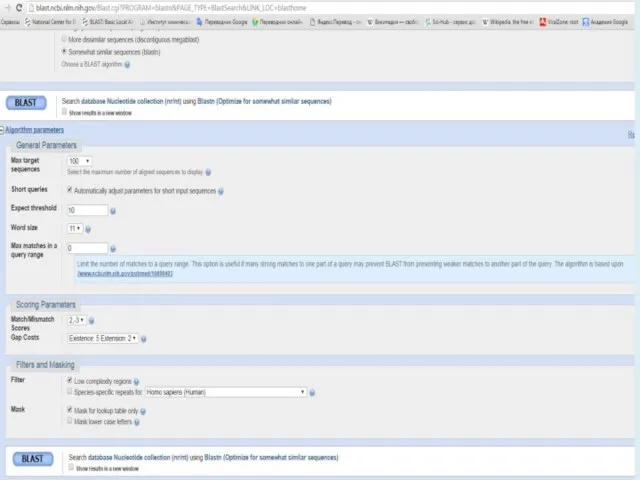
Выбор параметров
Меняйте параметры только, если по умолчанию не работает (параметры по
Выбор параметров
Меняйте параметры только, если по умолчанию не работает (параметры по

Выбор параметров
Матрица:BLOSUM для локального выравнивания обычно лучше, чем PAM
Чем выше номер
Выбор параметров
Матрица:BLOSUM для локального выравнивания обычно лучше, чем PAM
Чем выше номер



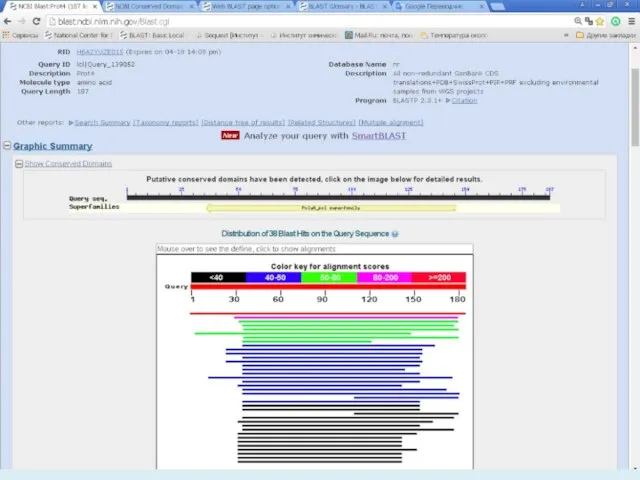
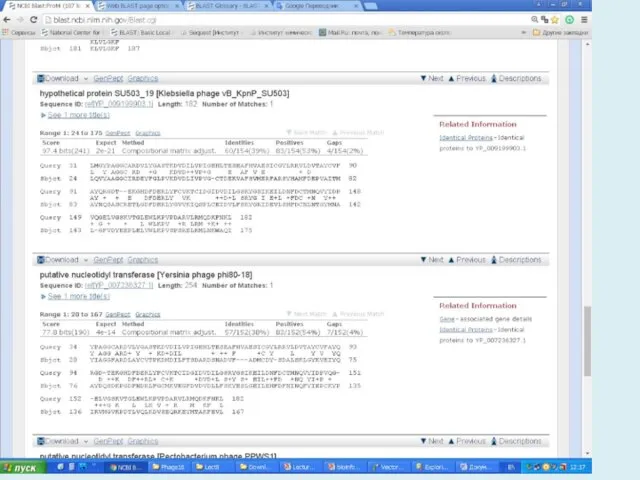
E-value (the expectation value) – представляет число различных элайментов с баллами
E-value (the expectation value) – представляет число различных элайментов с баллами



PSI – BLAST
Алгоритм:
Несколько раундов поиска
Первый раунд – просто blastp (BLOSUM62)
Построение
PSI – BLAST
Алгоритм:
Несколько раундов поиска
Первый раунд – просто blastp (BLOSUM62)
Построение


DELTA – BLAST (Domain Enhanced Lookup Time Accelerated BLAST)
DELTA – BLAST (Domain Enhanced Lookup Time Accelerated BLAST)



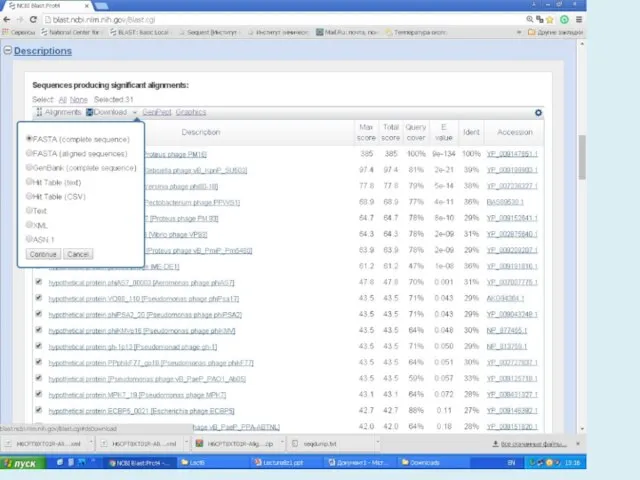



MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
Основное применение программы для разбора и анализа результата BLAST
MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
Основное применение программы для разбора и анализа результата BLAST

MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
The aim of MEGAN is to provide a tool
MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
The aim of MEGAN is to provide a tool

MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
However, the main application of the program is to
MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
However, the main application of the program is to

MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
New input to the program is usually provided as
MEGAN V5.11.3
http://ab.inf.uni-tuebingen.de/software/megan/
New input to the program is usually provided as


DNA Master
http://cobamide2.bio.pitt.edu/
DNA Master это Multiple Document Interface (MDI) программа для создания,
DNA Master
http://cobamide2.bio.pitt.edu/
DNA Master это Multiple Document Interface (MDI) программа для создания,

domain
A discrete portion of a protein assumed to fold independently of
domain
A discrete portion of a protein assumed to fold independently of

RAST (Rapid Annotation using Subsystem Technology) is a fully-automated service for annotating bacterial and archaeal genomes.
RAST (Rapid Annotation using Subsystem Technology) is a fully-automated service for annotating bacterial and archaeal genomes.


LOCUS FJ795028 701 bp mRNA linear PRI 06-APR-2009
DEFINITION Homo sapiens tumor
LOCUS FJ795028 701 bp mRNA linear PRI 06-APR-2009
DEFINITION Homo sapiens tumor

CDS <1..701
/gene="TNF"
/note="APC1 protein"
/codon_start=3
/product="tumor necrosis factor alpha"
/protein_id="ACO37640.1"
CDS <1..701
/gene="TNF"
/note="APC1 protein"
/codon_start=3
/product="tumor necrosis factor alpha"
/protein_id="ACO37640.1"
 Проводящие ткани
Проводящие ткани Происхождение растений. Основные этапы развития растительного мира
Происхождение растений. Основные этапы развития растительного мира Размножение та розвиток земноводних
Размножение та розвиток земноводних Чем вода полезна в жизни человека?
Чем вода полезна в жизни человека? Презентация на тему "Класс Млекопитающие или звери" - скачать презентации по Биологии
Презентация на тему "Класс Млекопитающие или звери" - скачать презентации по Биологии Теории происхождения человека
Теории происхождения человека Размножение декоративных растений. Классификация растений по декоративным качествам и направлениям использования в садоводстве
Размножение декоративных растений. Классификация растений по декоративным качествам и направлениям использования в садоводстве Строение головного мозга
Строение головного мозга Цветок - орган семенного размноженич
Цветок - орган семенного размноженич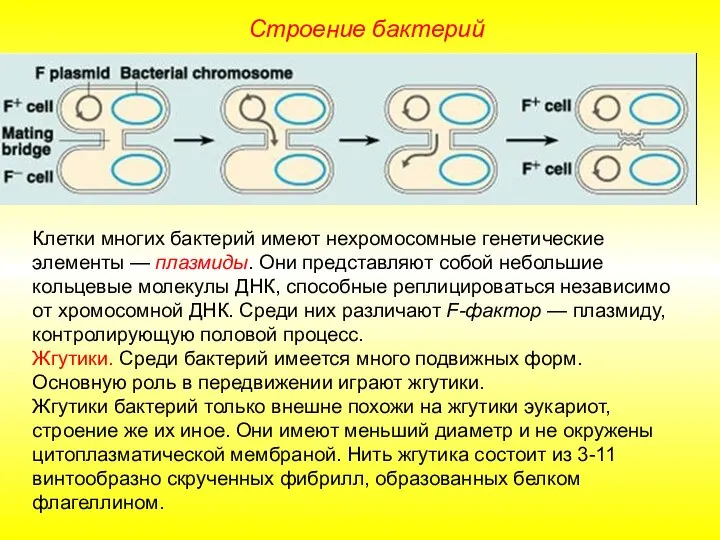 Презентация на тему "Строение бактерий" - скачать презентации по Биологии
Презентация на тему "Строение бактерий" - скачать презентации по Биологии Птицы. Птичьи разговоры
Птицы. Птичьи разговоры Митоз и мейоз
Митоз и мейоз Семейство розоцветные
Семейство розоцветные Учение Вернадского о биосфере
Учение Вернадского о биосфере Презентация на тему Плоскостопие
Презентация на тему Плоскостопие  Класс Млекопитающие (Звери)
Класс Млекопитающие (Звери) Теория филэмбриогенезов
Теория филэмбриогенезов Медузы
Медузы Хвороба Ебола Підготувала Учениця 11-А Безнощенко Валентина
Хвороба Ебола Підготувала Учениця 11-А Безнощенко Валентина  Методы размножения организмов
Методы размножения организмов Генетически-модифицированные организмы
Генетически-модифицированные организмы Population
Population Урок-практикум по решению задач по генетике
Урок-практикум по решению задач по генетике Культивирование и выделение чистых культур анаэробов
Культивирование и выделение чистых культур анаэробов Вплив різних видів фізичної активності на діяльність серця. Регуляція діяльності серця при фізичному навантаженні
Вплив різних видів фізичної активності на діяльність серця. Регуляція діяльності серця при фізичному навантаженні Разнообразие декоративных кустарников в моём городе
Разнообразие декоративных кустарников в моём городе Физиология микроорганизмов
Физиология микроорганизмов В гости к весне. Апрель
В гости к весне. Апрель