- Митохондриальные болезни

Содержание
- 2. МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ Гетерогенная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий
- 3. Каждая клетка организма содержит от нескольких сотен до тысяч органелл - митохондрий, содержащих от 2 до
- 4. Митохондриальная ДНК (мтДНК) человека содержит всего 37 генов; остальные примерно 25 тыс. генов, кодирующих белки, находятся
- 5. Каждая из митохондриальных хромосом кодирует 13 белков для 30 ферментов, ответственных за синтез АТФ, а также
- 6. Большая часть белков митохондрий (около 70) кодируется генами ядерной ДНК. Синтезируются эти полипептиды на рибосомах эндоплазматической
- 7. Особенности наследования МЗ Вследствие двойного кодирования компонентов мультиферментных комплексов, могут иметь любой тип наследования. Гетерогенность симптоматики
- 8. Одновременно в клетке могут сосуществовать нормальный (дикий) и мутантный типы мтДНК, что принято обозначать термином гетероплазмия.
- 9. Наследование мутаций в митохондриальном геноме носит особый характер. Если гены, заключенные в ядерной ДНК, дети получают
- 10. В составе митохондрий мтДНК наследуется по материнской линии
- 11. Человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной ДНК, соотношение митохондрий с мутантными
- 12. В подобных случаях мутации поначалу могут вообще не иметь внешних проявлений. Нормальные митохондрии до поры до
- 13. Однако рано или поздно наступает момент, когда дефектные формы накапливаются в количестве, достаточном для проявления патологических
- 14. Основная функция митохондрии, производство клеточной энергии, осуществляется дыхательной цепью. Дыхательная цепь локализуется во внутренней мембране митохондрии
- 15. Поступивший в цикл Кребса ацетил-СоА является конечным продуктом катаболизма углеводов, липидов и аминокислот (фенилаланин, тирозин, лейцин
- 16. Цикл Кребса (цикл лимонной кислоты) Цикл Кребса (цикл лимонной кислоты)
- 17. Окисленние липидов Митохондриальное окисление жирных кислот — главный источник энергии для сокращения миокарда, при голодании и
- 18. Карнитин (b-гидрокси-g-триметиламиномасляная кислота) — важный кофактор транспорта длинноцепочечных жирных кислот через митохондриальные мембраны. Дефекты трансмембранного переносчика
- 19. Карнитиновая кардиомиопатия Изолированная карнитиновая КМП клинически проявляется с 3-5 месяцев, имеет плохой прогноз, смерть наступает внезапно
- 20. Миопатический синдром определяется как прогрессирующая мышечная слабость, которая, за небольшими исключениями, проявляется в детстве. Отмечается слабость
- 21. Митохондриальное окисление длинноцепочечных жирных кислот последовательно осуществляют КПТ1 и 2 При дефекте гена CPT1 развивается печёночная
- 22. Острая энцефалопатия с отёком мозга и жировой инфильтрацией органов (преимущественно, печени), возникает у ранее здоровых новорождённых,
- 23. Механизм дыхательного фосфорилирования в митохондриях. Ферментные комплексы I, III и IV катализируют перенос электронов от НАДН
- 24. Частоту дисфункции дыхательной цепи оценивают от 1 на 5-10 тысяч до 4-5 на 100 тысяч новорожденных
- 25. Наиболее энергозависимыми, а потому уязвимыми являются мозг, сердце, скелетные мышцы, сенсорные органы, почечные канальцы, эндокринная система,
- 26. Первоначально МБ рассматривали как нервно-мышечную патологию или как митохондриальные энцефаломиелопатии. Нервно-мышечная патология обычно бывает представлена судорогами,
- 27. Сердечная патология при МБ в большинстве случаев представлена кардиомиопатией и дефектами проводимости, Эндокринопатии - гипогликемией и
- 28. Митохондриальная дисфункция может проявляться преимущественным поражением миокарда. Экспериментальными и клиническими исследованиями последних лет установлено, что при
- 29. Описаны КМП при дефиците цитохром С-оксидазы, снижении активности I и IV или II и III комплексов
- 30. Точная генетическая диагностика нарушений окислительного фосфорилирования затруднительна по причине выраженной генетической гетерогенности, клинического полиморфизма и семейного
- 31. Клиническая симптоматика, степень тяжести и исходы МП у детей варьируют в широких пределах. Главным прогностическим маркером
- 32. Синдром Пирсона ( Pearson syndrome ) Был описан Pearson в 1979 г. Характерными признаками считают: Упорную
- 34. МБ в основе мультиорганного патологического процесса при СП лежат делеции мтДНК. Фенотип и клиническое течение определяются
- 35. Большинство пациентов не достигают возраста 4-х лет. У пациентов, проживших несколько лет, в дальнейшем развиваются признаки
- 36. У большинства - вовлечение в патологический процесс поджелудочной железы, сочетающееся иногда с инсулинзависимым сахарным диабетом. Панкреатическая
- 37. В период новорожденности - гипотония, гипогликемия, тяжелый лактатацидоз в отсутствии анемии. Повышенное соотношение лактат/пируват в плазме
- 38. Лабораторное исследование лактатацидоз, комплексная органическая ацидурия, повышение содержания гемоглобина F и увеличение активности аденозиндезаминазы.
- 39. Биопсия скелетных мышц обнаруживает наличие характерных рваных красных волокон субсарколеманые скопления липидов, гликогена, кальция.
- 40. Митохондриальная нейрогастроинтестинальная энцефаломиопатия ( МНГИЭМ ) Мультисистемный синдром с вовлечением: Мышечной системы, Периферической и центральной нервной
- 41. Молекулярно-генетический анализ выявляет множественные делеции мтДНК и частичное истощение митохондриального генома. Ген, ассоциированный с МНГИЭМ и
- 42. МНГИЭМ наследуется аутосомно-рецессивно и относится к группе болезней, обозначаемых как дефекты межгеномного взаимодействия, нарушения мтДНК есть
- 43. Для заболевания характерны: Прогрессирующая внешняя офтальмоплегия (поражаются мышцы, осуществляющие движение глазного яблока), птоз, атрофия зрительного нерва,
- 44. Со стороны ЖК тракта отмечают тошноту, рвоту, боли, диарею, сниженную перистальтику кишечника. Хроническая псевдообструкция в сочетании
- 45. Исследование свежезамороженных скелетных мышц показывает наличие рваных красных волокон, аномальных митохондрий Полиморфные аномальные митохондрии среди миофибрилл.
- 46. Синдром Кернса-Сейра Впервые описан Кернсом в 1946 году. Более детальное изучение этого заболевания принадлежит Сейру в
- 47. При синдроме Кернса-Сейра при световой биопсии скелетных мышц феномен RRF определяется в 25% мышечных волокон. Обнаруживаются
- 48. Клиническая манифестация синдрома Кернса-Сейра относится ко второму или даже третьему десятилетию жизни. Этот феномен объясняется тем,
- 49. Отмечается задержка физического и полового развития. Изменения со стороны кожи проявляются ихтиозом с очагами гиперпигментации. Нарушения
- 50. Один из наиболее частых симптомов — мозжечковая атаксия. У многих больных наблюдается умственная отсталость, однако степень
- 51. Изменения сердечно-сосудистой системы являются облигатной составляющей клинического симптомокомплекса синдрома Кернса-Сейра. Варианты и степень выраженности нарушений со
- 52. Частота мутаций митохондриальной ДНК у пациентов с синдромом Кернса-Сейра в клетках проводящей системы составляет 35-40% по
- 53. Часто патология со стороны ССС при синдроме Кернса-Сейра долгое время остается нераспознанной. Появление полной АВ- блокады,
- 54. Синдром MELAS (митохондриальная миопатия — энцефалопатия — лактат-ацидоз - инсультоподобные эпизоды) Мутации митохондриальной ДНК наиболее часто
- 56. Экстракардиальные клинические симптомы MELAS синдрома Судороги, рецидивирующие головные боли, деменция, Инсультоподобные эпизоды, При компьютерной томографии головного
- 57. Cиндром MERRF (миоклонус-эпилепсия и инфаркт мозга, RRF-волокна) Сочетание миоклонус-эпилепсии с «рваными» красными волокнами скелетных мышц обнаружили
- 58. Синдром Барта (кардиомиопатия с нейтропенией и гипостатурой) В 1983 году П. Г. Барт и соавторы описали
- 59. Дети с данной патологией имеют низкий вес при рождении и в дальнейшем инфантильный соматотип (весо-ростовые показатели
- 60. Гистиоцитарная кардиомиопатия впервые была описана Д. Вотом в 1963 году . Клиническая симптоматика заболевания проявляется с
- 62. Скачать презентацию

МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Гетерогенная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий
МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Гетерогенная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий
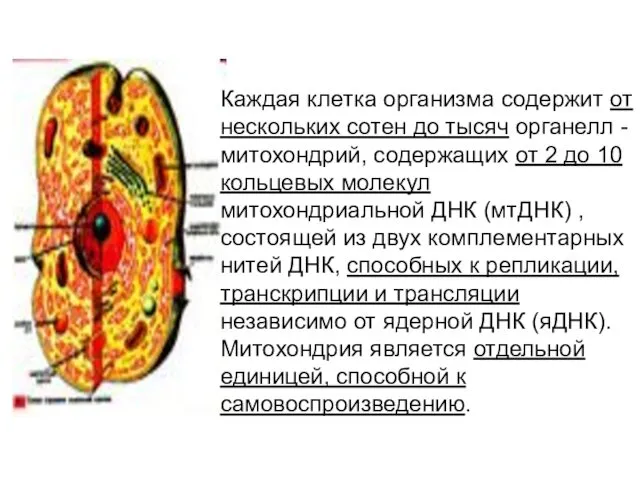
Каждая клетка организма содержит от нескольких сотен до тысяч органелл -
Каждая клетка организма содержит от нескольких сотен до тысяч органелл -

Митохондриальная ДНК (мтДНК) человека содержит всего 37 генов; остальные примерно
Митохондриальная ДНК (мтДНК) человека содержит всего 37 генов; остальные примерно
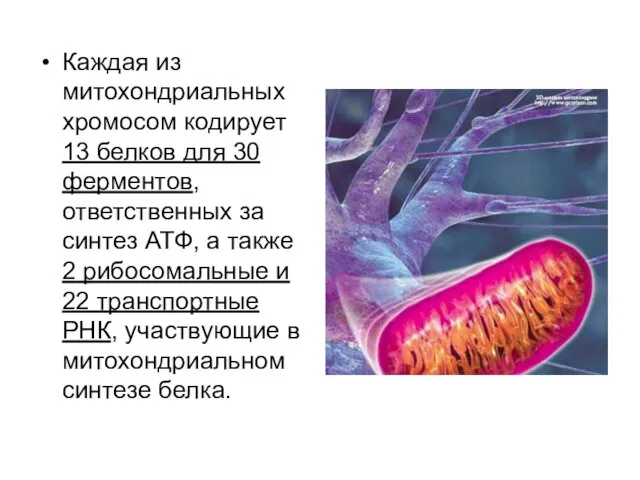
Каждая из митохондриальных хромосом кодирует 13 белков для 30 ферментов, ответственных
Каждая из митохондриальных хромосом кодирует 13 белков для 30 ферментов, ответственных

Большая часть белков митохондрий (около 70) кодируется генами ядерной ДНК.
Синтезируются
Большая часть белков митохондрий (около 70) кодируется генами ядерной ДНК.
Синтезируются

Особенности наследования МЗ
Вследствие двойного кодирования компонентов мультиферментных комплексов, могут иметь любой
Особенности наследования МЗ
Вследствие двойного кодирования компонентов мультиферментных комплексов, могут иметь любой

Одновременно в клетке могут сосуществовать нормальный (дикий) и мутантный типы мтДНК,
Одновременно в клетке могут сосуществовать нормальный (дикий) и мутантный типы мтДНК,

Наследование мутаций в митохондриальном геноме носит особый характер.
Если гены, заключенные
Наследование мутаций в митохондриальном геноме носит особый характер.
Если гены, заключенные

В составе митохондрий мтДНК наследуется по материнской линии
В составе митохондрий мтДНК наследуется по материнской линии
Человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной
Человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной
В подобных случаях мутации поначалу могут вообще не иметь внешних проявлений.
В подобных случаях мутации поначалу могут вообще не иметь внешних проявлений.

Однако рано или поздно наступает момент, когда дефектные формы накапливаются в
Однако рано или поздно наступает момент, когда дефектные формы накапливаются в

Основная функция митохондрии, производство клеточной энергии, осуществляется дыхательной цепью.
Дыхательная цепь
Основная функция митохондрии, производство клеточной энергии, осуществляется дыхательной цепью.
Дыхательная цепь

Поступивший в цикл Кребса ацетил-СоА является конечным продуктом катаболизма углеводов, липидов
Поступивший в цикл Кребса ацетил-СоА является конечным продуктом катаболизма углеводов, липидов
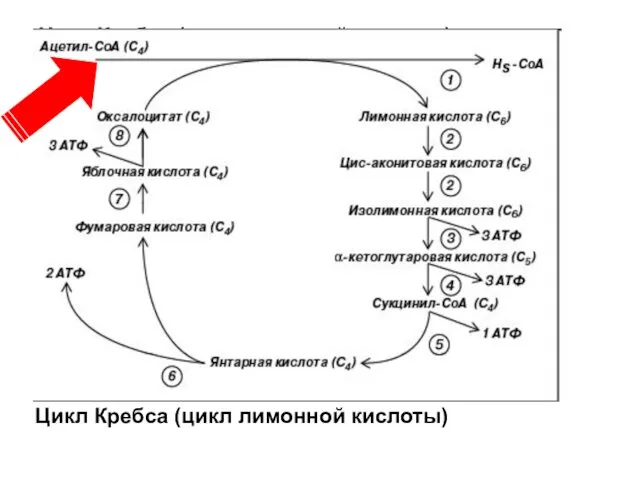
Цикл Кребса (цикл лимонной кислоты)
Цикл Кребса (цикл лимонной кислоты)
Цикл Кребса (цикл лимонной кислоты)
Цикл Кребса (цикл лимонной кислоты)
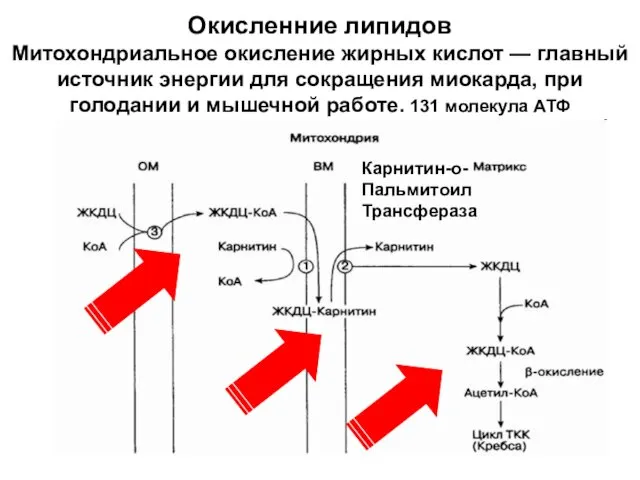
Окисленние липидов
Митохондриальное окисление жирных кислот — главный источник энергии для
Окисленние липидов Митохондриальное окисление жирных кислот — главный источник энергии для

Карнитин
(b-гидрокси-g-триметиламиномасляная кислота) — важный кофактор транспорта длинноцепочечных жирных кислот через
Карнитин (b-гидрокси-g-триметиламиномасляная кислота) — важный кофактор транспорта длинноцепочечных жирных кислот через

Карнитиновая кардиомиопатия
Изолированная карнитиновая КМП клинически проявляется с 3-5 месяцев, имеет плохой
Карнитиновая кардиомиопатия
Изолированная карнитиновая КМП клинически проявляется с 3-5 месяцев, имеет плохой

Миопатический синдром определяется как прогрессирующая мышечная слабость, которая, за небольшими исключениями,
Миопатический синдром определяется как прогрессирующая мышечная слабость, которая, за небольшими исключениями,

Митохондриальное окисление длинноцепочечных жирных кислот последовательно осуществляют КПТ1 и 2
При дефекте
Митохондриальное окисление длинноцепочечных жирных кислот последовательно осуществляют КПТ1 и 2
При дефекте

Острая энцефалопатия с отёком мозга и жировой инфильтрацией органов (преимущественно, печени),
Острая энцефалопатия с отёком мозга и жировой инфильтрацией органов (преимущественно, печени),
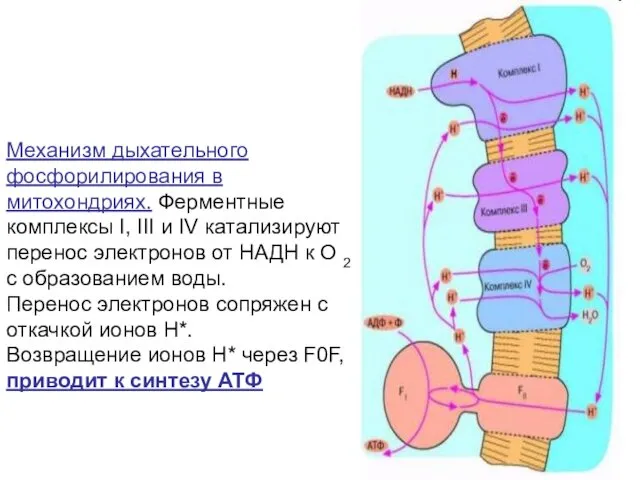
Механизм дыхательного фосфорилирования в митохондриях. Ферментные комплексы I, III и IV
Механизм дыхательного фосфорилирования в митохондриях. Ферментные комплексы I, III и IV

Частоту дисфункции дыхательной цепи оценивают от 1 на 5-10 тысяч до
Частоту дисфункции дыхательной цепи оценивают от 1 на 5-10 тысяч до
Наиболее энергозависимыми, а потому уязвимыми являются мозг, сердце, скелетные мышцы, сенсорные
Наиболее энергозависимыми, а потому уязвимыми являются мозг, сердце, скелетные мышцы, сенсорные

Первоначально МБ рассматривали как нервно-мышечную патологию или как митохондриальные энцефаломиелопатии. Нервно-мышечная
Первоначально МБ рассматривали как нервно-мышечную патологию или как митохондриальные энцефаломиелопатии. Нервно-мышечная

Сердечная патология при МБ в большинстве случаев представлена кардиомиопатией и дефектами
Сердечная патология при МБ в большинстве случаев представлена кардиомиопатией и дефектами
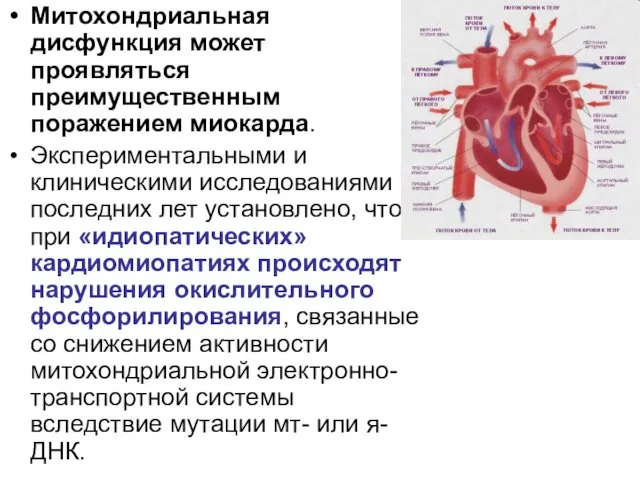
Митохондриальная дисфункция может проявляться преимущественным поражением миокарда.
Экспериментальными и клиническими исследованиями
Митохондриальная дисфункция может проявляться преимущественным поражением миокарда.
Экспериментальными и клиническими исследованиями

Описаны КМП при дефиците цитохром С-оксидазы, снижении активности I и IV
Описаны КМП при дефиците цитохром С-оксидазы, снижении активности I и IV

Точная генетическая диагностика нарушений окислительного фосфорилирования затруднительна по причине выраженной генетической
Точная генетическая диагностика нарушений окислительного фосфорилирования затруднительна по причине выраженной генетической

Клиническая симптоматика, степень тяжести и исходы МП у детей варьируют в
Клиническая симптоматика, степень тяжести и исходы МП у детей варьируют в

Синдром Пирсона
( Pearson syndrome )
Был описан Pearson в 1979 г. Характерными
Синдром Пирсона
( Pearson syndrome )
Был описан Pearson в 1979 г. Характерными
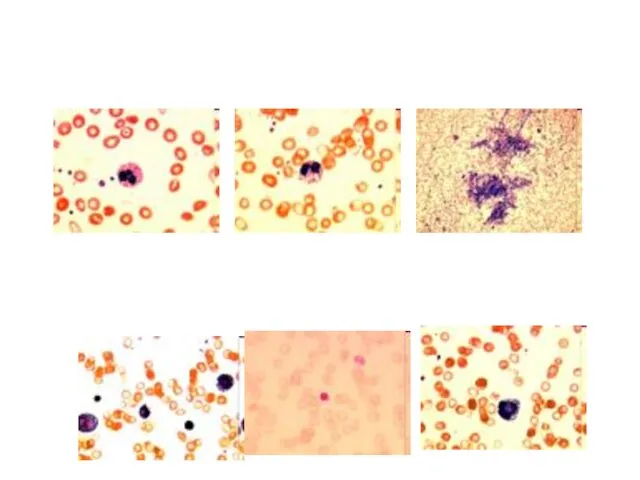

МБ в основе мультиорганного патологического процесса при СП лежат делеции мтДНК.
МБ в основе мультиорганного патологического процесса при СП лежат делеции мтДНК.

Большинство пациентов не достигают возраста 4-х лет. У пациентов, проживших несколько
Большинство пациентов не достигают возраста 4-х лет. У пациентов, проживших несколько
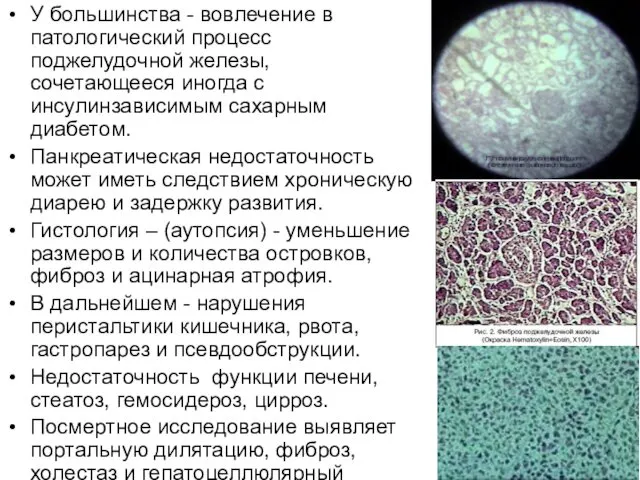
У большинства - вовлечение в патологический процесс поджелудочной железы, сочетающееся иногда
У большинства - вовлечение в патологический процесс поджелудочной железы, сочетающееся иногда

В период новорожденности - гипотония, гипогликемия, тяжелый лактатацидоз в отсутствии анемии.
В период новорожденности - гипотония, гипогликемия, тяжелый лактатацидоз в отсутствии анемии.

Лабораторное исследование
лактатацидоз,
комплексная органическая ацидурия, повышение содержания гемоглобина F и увеличение активности
Лабораторное исследование
лактатацидоз,
комплексная органическая ацидурия, повышение содержания гемоглобина F и увеличение активности

Биопсия скелетных мышц обнаруживает наличие характерных рваных красных волокон
субсарколеманые скопления липидов,
Биопсия скелетных мышц обнаруживает наличие характерных рваных красных волокон
субсарколеманые скопления липидов,
Митохондриальная нейрогастроинтестинальная энцефаломиопатия
( МНГИЭМ )
Мультисистемный синдром с вовлечением:
Мышечной системы,
Митохондриальная нейрогастроинтестинальная энцефаломиопатия
( МНГИЭМ )
Мультисистемный синдром с вовлечением:
Мышечной системы,

Молекулярно-генетический анализ выявляет множественные делеции мтДНК и частичное истощение митохондриального генома.
Ген,
Молекулярно-генетический анализ выявляет множественные делеции мтДНК и частичное истощение митохондриального генома.
Ген,

МНГИЭМ наследуется аутосомно-рецессивно и относится к группе болезней, обозначаемых как дефекты
МНГИЭМ наследуется аутосомно-рецессивно и относится к группе болезней, обозначаемых как дефекты
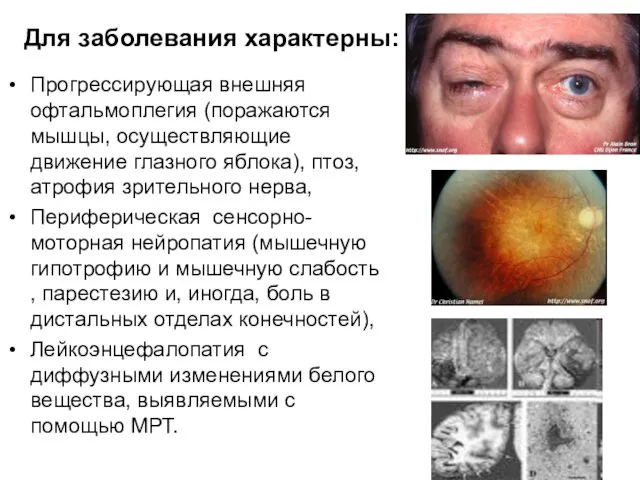
Для заболевания характерны:
Прогрессирующая внешняя офтальмоплегия (поражаются мышцы, осуществляющие движение глазного яблока),
Для заболевания характерны:
Прогрессирующая внешняя офтальмоплегия (поражаются мышцы, осуществляющие движение глазного яблока),

Со стороны ЖК тракта отмечают тошноту, рвоту, боли, диарею, сниженную перистальтику
Со стороны ЖК тракта отмечают тошноту, рвоту, боли, диарею, сниженную перистальтику
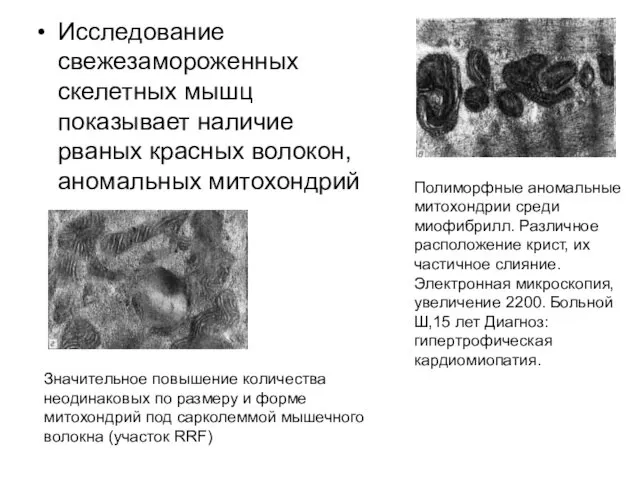
Исследование свежезамороженных скелетных мышц показывает наличие рваных красных волокон, аномальных митохондрий
Исследование свежезамороженных скелетных мышц показывает наличие рваных красных волокон, аномальных митохондрий
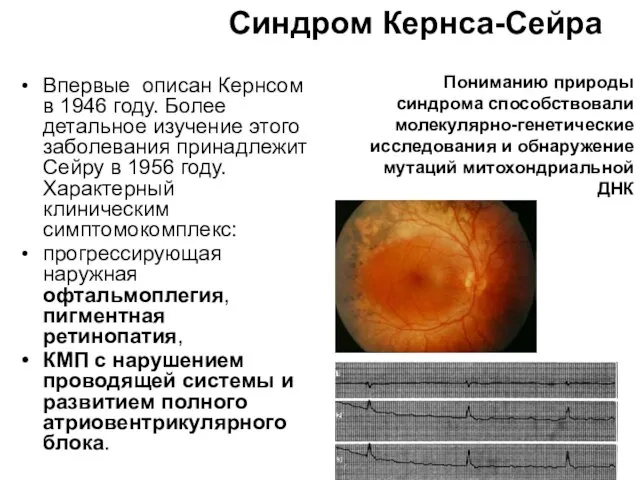
Синдром Кернса-Сейра
Впервые описан Кернсом в 1946 году. Более детальное изучение этого
Синдром Кернса-Сейра
Впервые описан Кернсом в 1946 году. Более детальное изучение этого
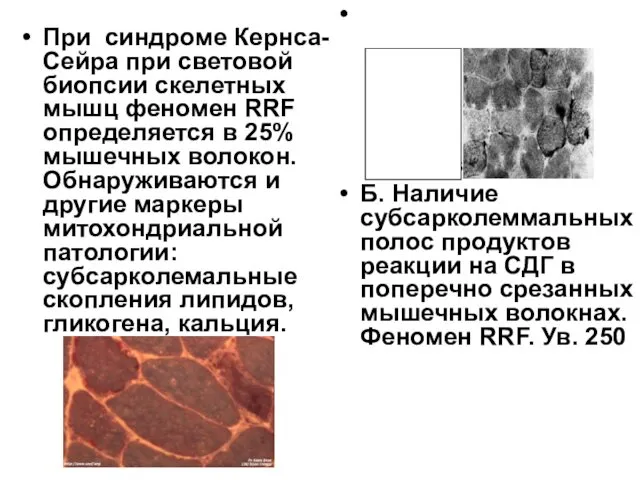
При синдроме Кернса-Сейра при световой биопсии скелетных мышц феномен RRF определяется
При синдроме Кернса-Сейра при световой биопсии скелетных мышц феномен RRF определяется

Клиническая манифестация синдрома Кернса-Сейра относится ко второму или даже третьему десятилетию
Клиническая манифестация синдрома Кернса-Сейра относится ко второму или даже третьему десятилетию
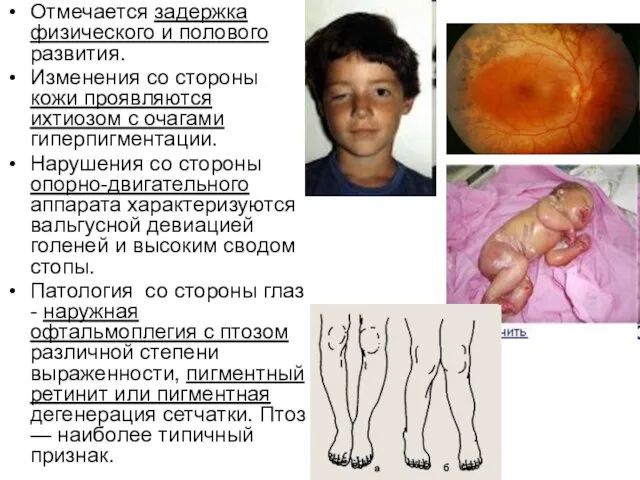
Отмечается задержка физического и полового развития.
Изменения со стороны кожи проявляются
Отмечается задержка физического и полового развития.
Изменения со стороны кожи проявляются
Один из наиболее частых симптомов — мозжечковая атаксия.
У многих больных
Один из наиболее частых симптомов — мозжечковая атаксия.
У многих больных
Изменения сердечно-сосудистой системы являются облигатной составляющей клинического симптомокомплекса синдрома Кернса-Сейра.
Варианты
Изменения сердечно-сосудистой системы являются облигатной составляющей клинического симптомокомплекса синдрома Кернса-Сейра.
Варианты
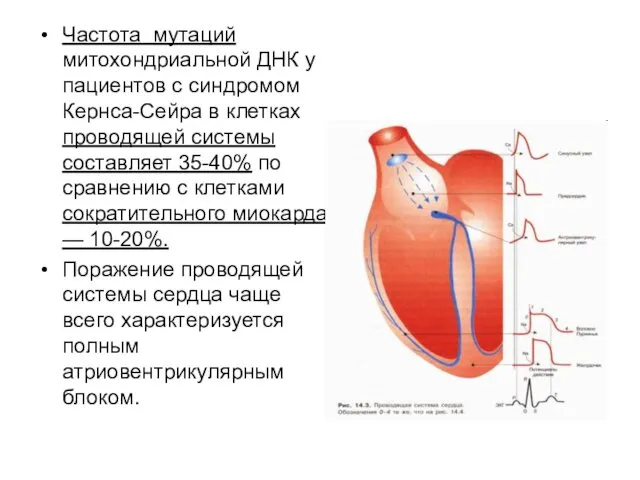
Частота мутаций митохондриальной ДНК у пациентов с синдромом Кернса-Сейра в клетках
Частота мутаций митохондриальной ДНК у пациентов с синдромом Кернса-Сейра в клетках

Часто патология со стороны ССС при синдроме Кернса-Сейра долгое время остается
Часто патология со стороны ССС при синдроме Кернса-Сейра долгое время остается

Синдром MELAS
(митохондриальная миопатия — энцефалопатия — лактат-ацидоз - инсультоподобные эпизоды)
Мутации
Синдром MELAS
(митохондриальная миопатия — энцефалопатия — лактат-ацидоз - инсультоподобные эпизоды)
Мутации
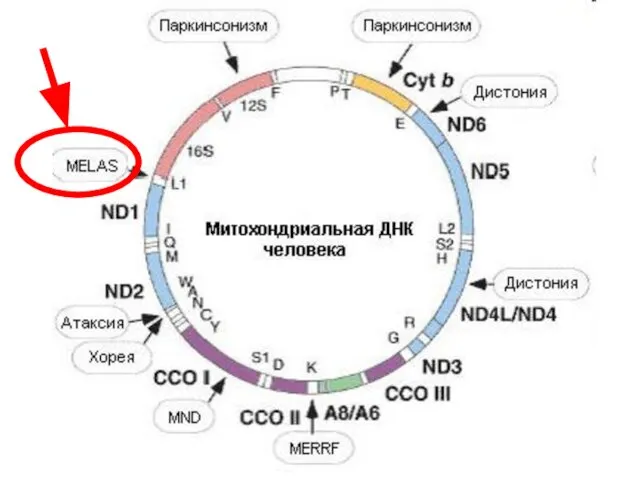
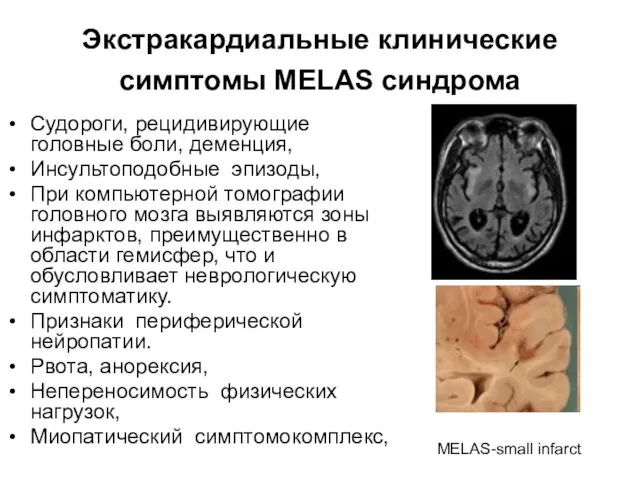
Экстракардиальные клинические симптомы MELAS синдрома
Судороги, рецидивирующие головные боли, деменция,
Инсультоподобные
Экстракардиальные клинические симптомы MELAS синдрома
Судороги, рецидивирующие головные боли, деменция,
Инсультоподобные

Cиндром MERRF
(миоклонус-эпилепсия и инфаркт мозга, RRF-волокна)
Сочетание миоклонус-эпилепсии с «рваными» красными волокнами
Cиндром MERRF
(миоклонус-эпилепсия и инфаркт мозга, RRF-волокна)
Сочетание миоклонус-эпилепсии с «рваными» красными волокнами

Синдром Барта
(кардиомиопатия с нейтропенией и гипостатурой)
В 1983 году П.
Синдром Барта
(кардиомиопатия с нейтропенией и гипостатурой)
В 1983 году П.

Дети с данной патологией имеют низкий вес при рождении и в
Дети с данной патологией имеют низкий вес при рождении и в

Гистиоцитарная кардиомиопатия
впервые была описана Д. Вотом в 1963 году .
Клиническая симптоматика
Гистиоцитарная кардиомиопатия
впервые была описана Д. Вотом в 1963 году .
Клиническая симптоматика
 Выделительная система. Строение и функции почек. Образование мочи
Выделительная система. Строение и функции почек. Образование мочи Туберкулез и его профилактика
Туберкулез и его профилактика Дифференциальная диагностика острого аппендицита с гинекологической и урологической патологией
Дифференциальная диагностика острого аппендицита с гинекологической и урологической патологией Аллергодерматозы у детей
Аллергодерматозы у детей Буллезный дерматоз. Пузырчатка
Буллезный дерматоз. Пузырчатка Анатомия грудной клетки
Анатомия грудной клетки Фитотерапия кожных заболеваний
Фитотерапия кожных заболеваний Тыныс жетіспеушілігі туралы жалпы түсінік
Тыныс жетіспеушілігі туралы жалпы түсінік Особенности психотерапевтической реабилитации пациентов с различными видами нехимических зависимостей
Особенности психотерапевтической реабилитации пациентов с различными видами нехимических зависимостей Психология личности
Психология личности Общий (гематологический) анализ крови
Общий (гематологический) анализ крови Гематология: лейкозы, геморрагические диатезы
Гематология: лейкозы, геморрагические диатезы Воля и волевые качества человека
Воля и волевые качества человека Емдік профилактикалық мекемелерде емдік дене шынықтыруды ұйымдастыру
Емдік профилактикалық мекемелерде емдік дене шынықтыруды ұйымдастыру Потребность пациента в нормальном дыхании
Потребность пациента в нормальном дыхании Сестринский персонал в программах профилактики ВИЧ. Лекция 1
Сестринский персонал в программах профилактики ВИЧ. Лекция 1 Вирусный гепатит D
Вирусный гепатит D Опухолевый рост
Опухолевый рост Systematization of grammar: "direct" and "indirect" speech. theme: Pathology of the form and structure of the teeth
Systematization of grammar: "direct" and "indirect" speech. theme: Pathology of the form and structure of the teeth Сучасний стан онкологічної служби в Україні. Організація онкологічної допомоги. Диспансеризація та облік онкологічних хворих
Сучасний стан онкологічної служби в Україні. Організація онкологічної допомоги. Диспансеризація та облік онкологічних хворих HIPEC при раке яичников
HIPEC при раке яичников Миокардит. Клинические признаки
Миокардит. Клинические признаки Исследование ликвора
Исследование ликвора Компенсаторно – приспособительные реакции
Компенсаторно – приспособительные реакции Поддерживающая терапия в период химиотерапии
Поддерживающая терапия в период химиотерапии Мариинская городская больница
Мариинская городская больница Возрастная анатомия, физиология и школьная гигиена
Возрастная анатомия, физиология и школьная гигиена Профилактическая медицина. Лекция № 1. История развития профилактики
Профилактическая медицина. Лекция № 1. История развития профилактики