- Моногенные наследственные болезни обмена веществ

Содержание
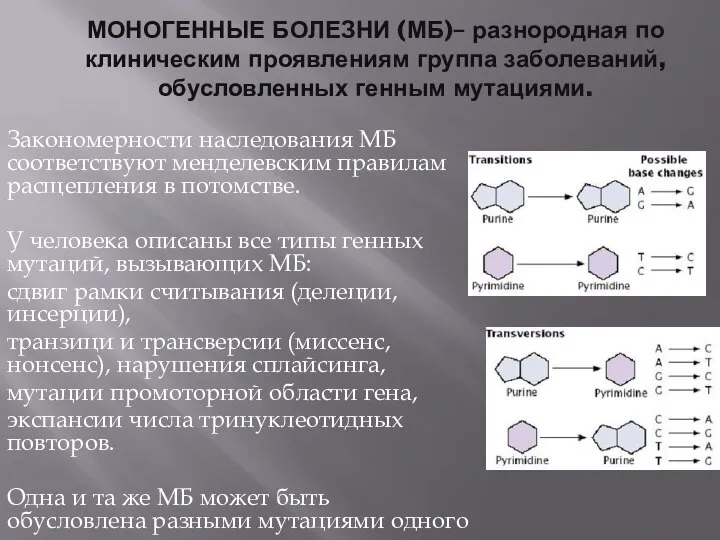
- 2. МОНОГЕННЫЕ БОЛЕЗНИ (МБ)– разнородная по клиническим проявлениям группа заболеваний, обусловленных генным мутациями. Закономерности наследования МБ соответствуют
- 3. Ген – структурно-функциональная единица наследственности, участок молекулы ДНК, кодирующий определенный белок или функциональную РНК. Гены, которые
- 4. Основная форма взаимодействия - полное доминирование, которое впервые описано Г. Менделем. В гетерозиготном организме проявление одной
- 5. Плейотропное действие генов - это зависимость нескольких признаков от одного гена, то есть множественное действие одного
- 6. Клинико-генетические базы данных Online Mendelian Inheritance in Man – OMIM www. Omim.org (Менделевское наследование у человека)
- 7. Классификация наследственных болезней обмена веществ: 1) Болезни углеводного обмена: Галактоземия (мутации гена галактозо-1-фосфатуридилтрансферазы, 9р13, ЧВ=1:15 000)
- 8. 9) Пероксисомные болезни: синдром Цельвегера (мутации в генах пероксинов, ЧВ=1:50 000) синдром Рефсума (мутации в гене
- 9. В России проводится массовый скрининг новорожденных (на 4 сутки после рождения (у недоношенных на 7) натощак
- 10. Орфанные болезни (ОБ) – редкие, затрагивающие небольшую часть популяции наследственные заболевания В США – ОБ –
- 11. БОЛЕЗНИ ОБМЕНА УГЛЕВОДОВ Углеводами называют органические вещества, которые содержат в своем составе карбонильную (=С=О) группу и
- 12. ГАЛАКТОЗЕМИЯ (E74.2). Впервые описана в 1908 г. А. Рессом. АР тип наследования, частота1:15 000. Дефицит ферментов,
- 13. ГАЛАКТОЗЕМИЯ (E74.2) ПАТОГЕНЕЗ Токсическое действие накапливающейся в результате метаболического блока галактозы и галактозо-1-фосфата на клетки ЦНС,
- 14. ГАЛАКТОЗЕМИЯ (E74.2) ДИАГНОСТИКА По результатам неонатального скрининга при повышении галактозы и снижении активности фермента галактозо-1-фосфатуридилтрансферазы. Если
- 15. ГЛИКОГЕНОЗЫ (Е74.0)
- 16. ГЛИКОГЕНОЗЫ (Е74.0) Возраст манифестации гликогенозов варьирует в зависимости от типа, но даже внутри одного типа бывают
- 17. ФРУКТОЗЕМИЯ Е 74.1 Ген ALDOB, 9q31.1, АЛЬДОЛАЗА-В. ЧВ = 1:20 000 новорожденных. Альдолаза В метаболизирует фруктозо-1фосфат
- 18. БОЛЕЗНИ ОБМЕНА АМИНОКИСЛОТ (АМИНОАЦИДОПАТИИ) Все аминоацидопатии подразделяются по МКБ-10 на следующие варианты: Е70 – Нарушения обмена
- 19. ФЕНИЛКЕТОНУРИЯ (E70.0, E70.1) ФКУ – АР тип наследования. Мутация гена РАН (фенилаланин-гидроксилаза), локализованного на 12q22-24. ЧВ=1:10
- 20. ФЕНИЛКЕТОНУРИЯ (E70.0, E70.1) В основе патогенеза ФКУ лежат аминоацидурия и ацидоз тканей, причинами которых являются следующие
- 21. Схема превращений фенилаланина и тирозина и основные метаболические блоки на их пути 1 - фенилкетонурия 2
- 22. ФЕНИЛКЕТОНУРИЯ (ФКУ) КЛИНИКА Первые проявления болезни: вялость, отсутствие интереса к окружающему, повышенная возбудимость, беспокойство, частое срыгивание,
- 23. ФЕНИЛКЕТОНУРИЯ (ФКУ) Диагностика БХ (превышение уровня ФА в крови более 900-1200 мкмоль/л); положительная проба Феллинга. Прямая
- 24. ТИРОЗИНЕМИЯ (E70.2) ЧВ=1:100 000. I тип - гепаторенальная – дефицит фермента фумарил-ацетоацетат-гидролазы (ген FAH, 15q23). Накапливаются
- 25. ТИРОЗИНЕМИЯ (E70.2) 2тип – глазокожная тирозинемия –дефицит фермента тирозинаминотрансферазы (ген TAT, 16q22.2). Клиника вследствие отложения кристаллов
- 26. ТИРОЗИНЕМИЯ (E70.2) 3 тип дефицит фермента 4гидроксифенил-пируватдиоксигеназы (HPD, 12q24.31). Характеризуется мягкой гипертирозинемией. Описаны бессимптомные формы и
- 27. ЛЕЙЦИНОЗ (болезнь «кленового сиропа») (E71.0) АР тип наследования, ЧВ=1:120 000. ЭТИОЛОГИЯ: Мутации в генах мультиферментного комплекса
- 28. ЛЕЙЦИНОЗ (болезнь «кленового сиропа») (E71.0) Клиника: 4 варианта течения (при всех будет УО): 1) Классический –
- 29. ГОМОЦИСТИНУРИЯ (E72.1) Нарушение обмена аминокислоты метионина. АР тип наследования, ген 21q22.1. ЧВ=1:200 000. Типы: I (цистатионин-бета-синтаза,
- 30. ГОМОЦИСТИНУРИЯ (E72.1) Диагностика: БХ анализ крови и мочи (высокое содержание гомоцистина и метионина при низком уровне
- 31. АЛКАПТОНУРИЯ (Е 70.2) Впервые описано Арчибальдом Гарродом в 1909 году. Мутация в гене оксизады гомогентезиновой кислоты
- 32. Дефицит ацил-КоА-дегидрогеназы (Е71.3) ЧВ=1:50 000. Мутации в гене VLCAD (17p.13). Синоним гена ACADVL. Клинические формы: Системная
- 33. Болезнь Леша-Нихена (Е79.1) ЧВ=1:300 000. (В 1964 г. – Майкл Лёш и Билл Нихен). Мутации гена
- 34. Болезнь Леша-Нихена (Е79.1) Клиника: хореоатетоз (хорея – быстрые прерывистые движения, атетоз – медленные судорожные движения), задержка
- 35. БОЛЕЗНИ ОБМЕНА ПОРФИРИНОВ Порфирины – предшественники гема – образуются в ходе его синтеза из глицина и
- 36. Острая перемежающаяся порфирия (Е80.2) ЧВ=1:100 000 населения. АД тип наследования, неполная пенетрантность. Мутация гена третьего фермента
- 37. МУКОВИСЦЕДОЗ (E84.0) Мутация в гене муковисцедозного трансмембранного регулятора проводимости, 7q31-32. ЧВ=1:5000. Ген экспрессируется в эптелии, регулирует
- 38. БОЛЕЗНИ ОБМЕНА ГОРМОНОВ Строение гонана (циклопентан-пергидрофенантрена), основы строения стероидов
- 39. АДРЕНО-ГЕНИТАЛЬНЫЙ СИНДРОМ (Е25) Мутация в гене стероид-21-гидроксилазы (CYP21А2), локализация 6р21.3. ЧВ=1:15000. Фермент стероид-21-гидроксилаза необходим для синтеза
- 40. АДРЕНО-ГЕНИТАЛЬНЫЙ СИНДРОМ (Е25)
- 41. ВРОЖДЕННЫЙ ГИПОТИРЕОЗ (Е00) Мутации в генах TITF1 (тиреоидный фактор транскрипции), локализация 14q13-21. ЧВ=1:5000. Крупный плод, отек
- 42. ВРОЖДЕННЫЙ ГИПОТИРЕОЗ (Е00) Диагностический меркер – тироксин, ТТГ крови. В норме у новорожденных уровень тироксина =22
- 43. БОЛЕЗНЬ ВИЛЬСОНА-КОНОВАЛОВА (Е83.0) Заболевание обусловлено мутациями в гене АТР7В (13q14.3). Ген кодирует Cu-зависимую АТФ-азу (печеночную). АР
- 44. Патогномоничным симптомом заболевания является зеленовато-бурое кольцо отложений меди по периферии радужной оболочки (кольцо Кайзера-Флейшера), обнаруживаемое при
- 45. БОЛЕЗНЬ МЕНКЕСА (Е83.0) Ген АТР7А (Xq21.1). Ген кодирует Cu-зависимую АТФ-азу (кишечную), в результате в кишечнике не
- 46. БОЛЕЗНЬ МЕНКЕСА (Е83.0) Синдром «затылочного рога» проявляется в ювенильном или взрослом возрасте в виде повышенной растяжимости
- 47. ГЕМОХРОМАТОЗ (Е83.1) Накопление избытка Fe в органах, c их тяжелым поражением (цирроз, диабет, кардиомиопатия, артрит). ЧВ=1:300.
- 48. Синдром Кернса-Сейра (H49.8) Делеци в мтДНК. Данные о ЧВ не представлены. Клинически: Дебют в 4 –
- 49. Синдром MELAS (G71.3) Митохондриальная энцефалопатия, лактоацидоз и инсультоподобные эпизоды. ЧВ=1:10 000 человек. Мутации в мтДНК: гены:
- 50. Синдром MERRF (G71.3) Миоклоническая эпилепсия с рваными мышечными волокнами. Мутации в мтДНК: MTTK, MTTL1, MTTH, MTTS1,
- 51. Мукополисахаридозы Мукополисахаридозы – группа заболеваний, обусловленных генетическим дефектом ферментного расщепления углеводной части молекулы мукополисахаридов (кислые ГАГ,
- 52. МУКОПОЛИСАХАРИДОЗЫ МПС
- 53. МПС I типа – синдром Гурлера (Е76.0) ЧВ= 1:100 000 населения. АР т/н. Ген IDUA, 4p16.3.
- 54. Рентгенологические изменения при синдроме Гурлер: Кубовидной формы позвонки с закругленными контурами, углообразный кифоз пояснично-грудного отдела, гипоплазия
- 55. МПС I типа – синдром Шейе (Е76.0) Пациенты с вариантом Шейе имеют нормальный или почти нормальный
- 56. СИНДРОМ ГУРЛЕРА-ШЕЙЕ (Е76.0) симптомы поражения скелета и внутренних органов разной степени на фоне нормального или почти
- 57. МПС I типа Диагностика: биохимический метод (повышение в моче уровня дерматансульфатов и гепарансульфатов и определение активности
- 58. Синдром Хантера (Е76.1) ЧВ= 1:132 000 населения. Ген IDS, Xq28. ИДУРОНАТ-L-СУЛЬФАТАЗА КП: когнитивные расстройства, микрогнатия, грубые
- 59. Синдром Санфилиппо (Е76.2) Тип А (1:100 000, SGSH, 17q25.3, СУЛЬФОГИДРОЛАЗА N-СУЛЬФОГЛЮКОЗАМИНА), тип B (1:200 000, NAGLU,
- 60. Синдром Моркио (Е76.2) Тип А (ЧВ=1:40 000, АР т/н, ген GALNS, 16q24.3, ГАЛАКТОЗАМИН-6-СУЛЬФАТ СУЛЬФАТАЗА). Тип В
- 61. СИНДРОМ МАРОТО-ЛАМИ (Е76.2) ЧВ=1:215 000. АР т/н. Ген ARSB, 5q14.1. АРИЛСУЛЬФАТАЗА-В. Накопление в организме дерматансульфата. Клиника:
- 62. СИНДРОМ СЛАЯ (Е76.2) ЧВ= 1:1 250 000. АР т/н. Ген GUSB, 7q11.21. БЕТА-ГЛЮКУРОНИДАЗА Клинка: грубые черты
- 63. Синдром Натовича (Е76.2) ЧВ неизвестна. АР т/н. Ген HYAL1, 3p21.31. ГЛЮКОЗАМИНИДАЗУ ГИАЛУРОНОВОЙ КИСЛОТЫ Клиника ювенильного идеопатического
- 64. СФИНГОЛИПИДОЗЫ При сфинголипидозах – внутриклеточное накопление сфинголипидов в головном и костном мозге, легких, печени и селезенке.
- 65. Niemann-Pick diseases (Е75.2) ЧВ типа А и В = 1:40 000, типа С = 1:100 000.
- 66. Fabry disease (Е75.2) ЧВ=1:40 000. Ген GLA, Xq22.1 (альфа-галактозидаза А). Накопление глоботриаозилцерамида. Клиника: грубые черты лица.
- 67. GM1-ГАНГЛИОЗИДОЗ (БОЛЕЗНЬ ТЕЯ-САКСА) (E75.0) АР тип наследования. Ген HEXA, 15q23, альфа субъединица гексаминидазы. ЧВ=1:320000, еври-ашкеназы 1:3500.
- 68. Gaucher’s disease(E75.2) Ген GBA, 1q22. БЕТА-ГЛЮКОЦЕРЕБРОЗИДАЗА. Накопление глюкоцереброзида – клетки Гоше. Тип I (ненейронопатический) –1:50 000,
- 69. БГ тип 1 до и после ферментзаместительной терапии Девочка 11 мес, БГ тип 2 Мальчик 3
- 70. БОЛЕЗНЬ КРАББЕ (E75.2) ЧВ=1:100 000, ген GALC, 14q31. ГАЛАКТОЦЕРЕБРОЗИДАЗА. Накопление в ЦНС галактозилерамидов. Дебют в 3
- 71. ПЕРОКСИСОМНЫЕ БОЛЕЗНИ Пероксисома - обязательная органелла эукариотической клетки, ограниченная мембраной, содержащая от 15 до 50 различных
- 72. 1. Пероксисомы получили такое название благодаря тому, что обычно в их состав входит один или несколько
- 73. В пероксисомах происходит окисление длинноцепочечных жирных кислот с образованием ацил-CoA, который переходит в цитозоль для повторного
- 74. АДРЕНОЛЕЙКОДИСТРОФИЯ (Е71.3) ЧВ=1:20 000. Ген ABCD1, Xq28. Кодирует транспортер жирных кислот с очень длинной цепью (ALD).
- 75. Refsum disease (G60.1) Ген PHYH, 6q22 – ФИТАНОИЛ-КОА-ГИДРОКСИЛАЗА. ЧВ=1:25 000. Накопление фитановой кислоты в в ЦНС,
- 76. СИНДРОМ ЦЕЛЬВЕГЕРА (Q87.8) АР т/н. ЧВ=1:50 000. Гены PEX1, 2, 3, 5, 6, 10, 12. 60%
- 77. Синдром Цельвегера Скафоцефалия (преждевременное зарастание швов черепа). Гипоплазия носа. Выпуклый куполообразный лоб. Низкопосаженные уши. Ретрогнатия (сдвиг
- 78. Принципы патогенетического лечения НБОВ: Устранение поступления субстрата с пищей. Коррекция выведения продукта (болезнь Леша-Нихена – аллопуринол,
- 79. Гемофилии D66 Гемофилия А. Ген VIII п.ф.с., Xq28, ЧВ=1:10 000. Гемофилия В. Ген F9, Xq27, ЧВ=1:50
- 80. НАСЛЕДСТВЕННЫЕ ДИСЛИПИДЕМИИ 1 тип – гиперхиломикронемия (ген липопротеинлипазы- 8p22, ген апоС2 – 19q32), ЧВ=1:1000. 2а тип
- 81. Наследственные иммунодефициты. Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1 : 25000 до 1
- 82. ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов – B+ТКИД и B-ТКИД. В
- 83. АДА-дефицит составляет около 15% всех случаев ТКИД и около трети – его аутосомно-рецессивных форм. Клинический полиморфизм
- 84. В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что генетическая конструкция, содержащая нормальный ген
- 85. 1) по типу клеток-мишеней: соматическая фетальная 2) по цели воздействия: позитивная ( компенсация экспресcии гена) негативная
- 86. Вирусные векторы: ретровирусы аденовирусы аденоассоциированный вирус герпесвирусы лентивирусы и др. - достоинства: трансфекция большого количества клеток
- 87. Аденовирусы Преимущества: способны инфицировать неделящиеся клетки большая клонирующая емкость (в настоящее время - до 28 т.п.о.)
- 88. Ретровирусы Преимущества: не иммуногенны постоянная экспрессия целевых генов Недостатки: инфицируют только делящиеся клетки потенциальная туморогенность низкий
- 89. Невирусные системы прямая инъекция рецепторо-опосредованный эндоцитоз генное ружье липофекция электропорация полимерные носители
- 90. Плазмидные векторы - достоинства: отсутствие токсичности и мутагенности практически неограниченная емкость вектора дешевизна производства - недостатки:
- 91. Клинические испытания генотерапевтических препаратов. I фаза. Оценка токсичности генной конструкции. II фаза. Ограниченные испытания на небольшом
- 92. ПРИМЕРЫ: муковисцидоз (кистозный фиброз поджелудочной железы) - перенос гена МТР (муковисцидозный трансмембранный регулятор) с помощью аденовирусного
- 95. WBC (white blood cells) = 4 – 9 x 109 /л RBC (red blood cells) =
- 96. По степени тяжести: легкой ( Hb >90 г/л), средней (70 – 90) и тяжелой (менее 70
- 97. Гемолитическая анемия Гемолитическая анемия возникает при преобладании процесса разрушения эритроцитов над их образованием. Наследственные гемолитические анемии
- 98. Апластическая анемия Фалькони . Гипоплатическая анемия Ерлиха. Анемия Даймонда-Блекфена. Синдром Швахмана-Даймонда. Наследственные апластические анемии
- 99. МЕМБРАНОПАТИИ сфероцитоз Минковского-Шоффара наследственный элиптоцитоз наследственный пиропойкилоцитоз наследственный стоматоцитоз наследственный акантоцитоз наследственный эхиноцитоз
- 100. АД тип наследования. ЧВ= 1 : 5000 населения. Около 25% случаев спорадические. Мутации гена спектрина мембраны
- 101. манифестация в раннем детском возрасте ДИАГНОСТИКА: -наследственный анамнез. -осмотр (башенный череп, готическое небо, изменения расположения зубов,
- 103. Наследственный сфероцитоз Лечение: Бессимптомные формы Лечения не требуется УЗИ контроль состояния желчных путей Легкая и среднетяжелая
- 104. Ферментопатии Недостаточность эритроцитарных ферментов: глюкозо-6-фосфатдегидрогеназа пируваткиназа глюкозофосфат изомераза
- 105. ген - на Х-хромосоме (Xq28) – наследование Х-сцепленное, болеют мальчики; редко девочки -гомозигы Девочки-гетерозиготы имеют две
- 106. острая гемолитическая анемия развивается спустя несколько часов или дней от начала приема лекарств, контакта с нафталином,
- 107. Дефицит глюкозо-6-фосфат дегидрогеназы (Г-6-ФД) Диагностика: Признаки внутрисосудистого гемолиза – увеличение непрямого билирубина, ЛДГ, свободного гемоглобина плазмы,
- 108. Серповидно-клеточная анемия АР тип наследования. В норме гемоглобин человека состоит из 2 альфа-цепей и двух бета-цепей.
- 109. ГЕМОГЛОБИНОПАТИИ Серповидноклеточная анемия. У таких больных вместо гемоглобина А синтезируется гемоглобин S. Отличается он тем, что
- 110. Серповидноклеточная анемия. Клинические проявления. Конституциональные проявления – отставание роста и развития, полового созревания. 2. Повышенная склонность
- 111. Серповидноклеточная анемия. Клинические проявления. Апластические кризы. Пациенты с СКА подвержены инфекциям и воспалительным процессам, подавляющим эритропоэз.
- 112. Серповидноклеточная анемия. Критерии диагноза. 1. Изменения со стороны анализа крови: макроцитоз эритроцитов (MCV>100), ретикулоцитоз (>100,0*10^9/л), лейкоцитоз
- 113. Серповидно-клеточная анемия Терапия Инфузионная терапия, анальгетики Трансфузионная терапия – только по показаниям (с осторожностью!!!, возможно увеличение
- 114. ТАЛАССЕМИЯ Это группа аутосомно – рецессивных заболеваний крови, характеризующихся снижением синтеза одного из двух полипептидных цепей
- 115. Молекулярные основы талассемий. Синтез a – цепей глобина кодируется диплоидным набором генов aa/aa (4 гена, по
- 116. Молекулярные основы талассемий. Малая талассемия (В+/В) протекает, как правило, без клинических проявлений, за исключением более высокой
- 117. Талассемии. Клинические проявления. 1) Анемия хронического течения, вызывающая замедление роста, полового развития, хроническую сердечную недостаточность и
- 118. Талассемии. Критерии диагноза. А. В анализе крови анемия различной степени, микроцитоз эритроцитов (MCV Б. Наличие симптомов
- 119. Талассемии. Лечение. При тяжёлых формах: 1) Регулярные трансфузии эритроцитарной массы (1 – 3 дозы каждые 3
- 121. Скачать презентацию
МОНОГЕННЫЕ БОЛЕЗНИ (МБ)– разнородная по клиническим проявлениям группа заболеваний, обусловленных генным
МОНОГЕННЫЕ БОЛЕЗНИ (МБ)– разнородная по клиническим проявлениям группа заболеваний, обусловленных генным

Ген – структурно-функциональная единица наследственности, участок молекулы ДНК, кодирующий определенный белок
Ген – структурно-функциональная единица наследственности, участок молекулы ДНК, кодирующий определенный белок

Основная форма взаимодействия - полное доминирование, которое впервые описано Г. Менделем.

Плейотропное действие генов - это зависимость нескольких признаков от одного гена,
Плейотропное действие генов - это зависимость нескольких признаков от одного гена,

Клинико-генетические базы данных
Online Mendelian Inheritance in Man – OMIM
www. Omim.org (Менделевское
Клинико-генетические базы данных
Online Mendelian Inheritance in Man – OMIM
www. Omim.org (Менделевское

Классификация наследственных болезней обмена веществ:
1) Болезни углеводного обмена:
Галактоземия (мутации гена галактозо-1-фосфатуридилтрансферазы,
Классификация наследственных болезней обмена веществ:
1) Болезни углеводного обмена:
Галактоземия (мутации гена галактозо-1-фосфатуридилтрансферазы,

9) Пероксисомные болезни:
синдром Цельвегера (мутации в генах пероксинов, ЧВ=1:50 000)
синдром Рефсума
9) Пероксисомные болезни:
синдром Цельвегера (мутации в генах пероксинов, ЧВ=1:50 000)
синдром Рефсума
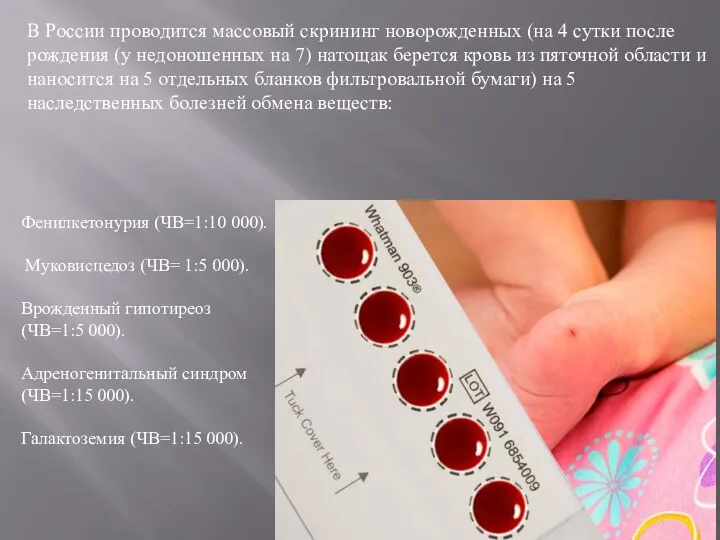
В России проводится массовый скрининг новорожденных (на 4 сутки после рождения
В России проводится массовый скрининг новорожденных (на 4 сутки после рождения

Орфанные болезни (ОБ) – редкие, затрагивающие небольшую часть популяции наследственные заболевания
В
Орфанные болезни (ОБ) – редкие, затрагивающие небольшую часть популяции наследственные заболевания
В

БОЛЕЗНИ ОБМЕНА УГЛЕВОДОВ
Углеводами называют органические вещества, которые содержат в своем составе
БОЛЕЗНИ ОБМЕНА УГЛЕВОДОВ
Углеводами называют органические вещества, которые содержат в своем составе
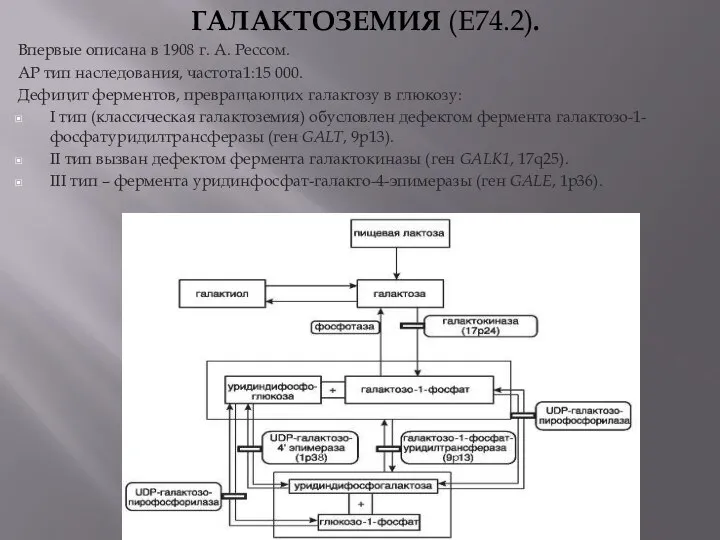
ГАЛАКТОЗЕМИЯ (E74.2).
Впервые описана в 1908 г. А. Рессом.
АР тип наследования,
ГАЛАКТОЗЕМИЯ (E74.2).
Впервые описана в 1908 г. А. Рессом.
АР тип наследования,
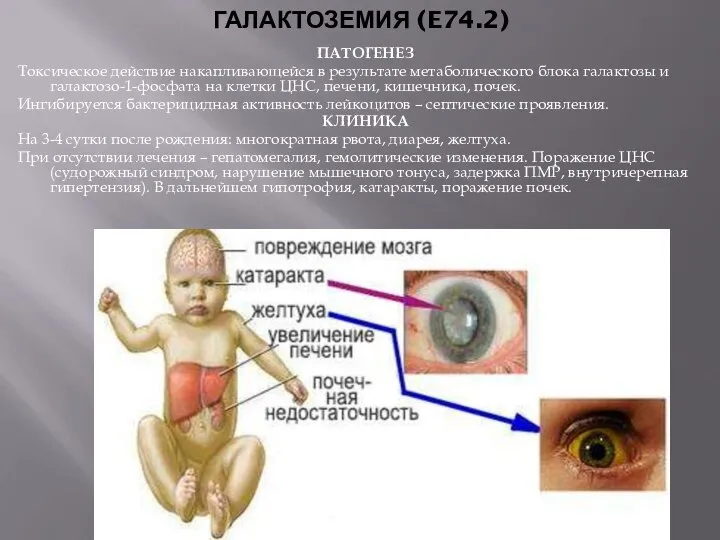
ГАЛАКТОЗЕМИЯ (E74.2)
ПАТОГЕНЕЗ
Токсическое действие накапливающейся в результате метаболического блока галактозы и галактозо-1-фосфата
ГАЛАКТОЗЕМИЯ (E74.2)
ПАТОГЕНЕЗ
Токсическое действие накапливающейся в результате метаболического блока галактозы и галактозо-1-фосфата

ГАЛАКТОЗЕМИЯ (E74.2)
ДИАГНОСТИКА
По результатам неонатального скрининга при повышении галактозы и снижении активности
ГАЛАКТОЗЕМИЯ (E74.2)
ДИАГНОСТИКА
По результатам неонатального скрининга при повышении галактозы и снижении активности

ГЛИКОГЕНОЗЫ (Е74.0)
ГЛИКОГЕНОЗЫ (Е74.0)

ГЛИКОГЕНОЗЫ (Е74.0)
Возраст манифестации гликогенозов варьирует в зависимости от типа, но даже
ГЛИКОГЕНОЗЫ (Е74.0)
Возраст манифестации гликогенозов варьирует в зависимости от типа, но даже

ФРУКТОЗЕМИЯ Е 74.1
Ген ALDOB, 9q31.1, АЛЬДОЛАЗА-В.
ЧВ = 1:20 000 новорожденных.
Альдолаза В
ФРУКТОЗЕМИЯ Е 74.1
Ген ALDOB, 9q31.1, АЛЬДОЛАЗА-В.
ЧВ = 1:20 000 новорожденных.
Альдолаза В
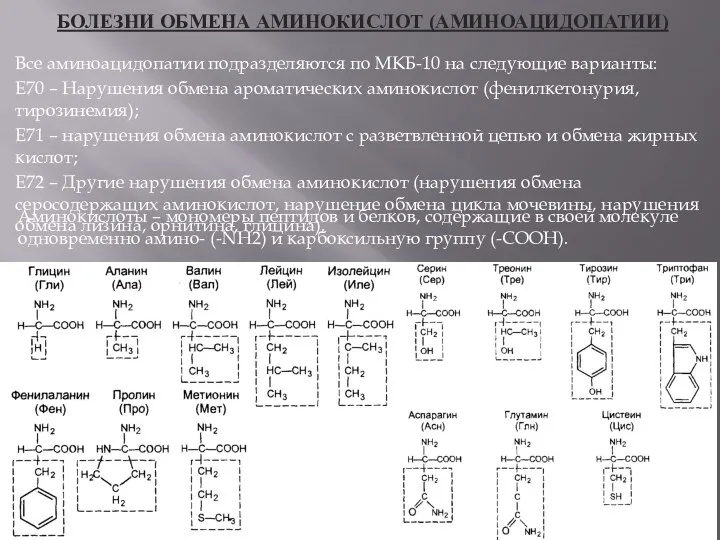
БОЛЕЗНИ ОБМЕНА АМИНОКИСЛОТ (АМИНОАЦИДОПАТИИ)
Все аминоацидопатии подразделяются по МКБ-10 на следующие варианты:
БОЛЕЗНИ ОБМЕНА АМИНОКИСЛОТ (АМИНОАЦИДОПАТИИ)
Все аминоацидопатии подразделяются по МКБ-10 на следующие варианты:

ФЕНИЛКЕТОНУРИЯ (E70.0, E70.1)
ФКУ – АР тип наследования. Мутация гена РАН (фенилаланин-гидроксилаза),
ФЕНИЛКЕТОНУРИЯ (E70.0, E70.1)
ФКУ – АР тип наследования. Мутация гена РАН (фенилаланин-гидроксилаза),

ФЕНИЛКЕТОНУРИЯ (E70.0, E70.1)
В основе патогенеза ФКУ лежат аминоацидурия и ацидоз тканей,
ФЕНИЛКЕТОНУРИЯ (E70.0, E70.1)
В основе патогенеза ФКУ лежат аминоацидурия и ацидоз тканей,

Схема превращений фенилаланина и тирозина и основные метаболические блоки на их
Схема превращений фенилаланина и тирозина и основные метаболические блоки на их

ФЕНИЛКЕТОНУРИЯ (ФКУ)
КЛИНИКА
Первые проявления болезни: вялость, отсутствие интереса к окружающему, повышенная
ФЕНИЛКЕТОНУРИЯ (ФКУ)
КЛИНИКА
Первые проявления болезни: вялость, отсутствие интереса к окружающему, повышенная

ФЕНИЛКЕТОНУРИЯ (ФКУ)
Диагностика
БХ (превышение уровня ФА в крови более 900-1200 мкмоль/л);
ФЕНИЛКЕТОНУРИЯ (ФКУ)
Диагностика
БХ (превышение уровня ФА в крови более 900-1200 мкмоль/л);
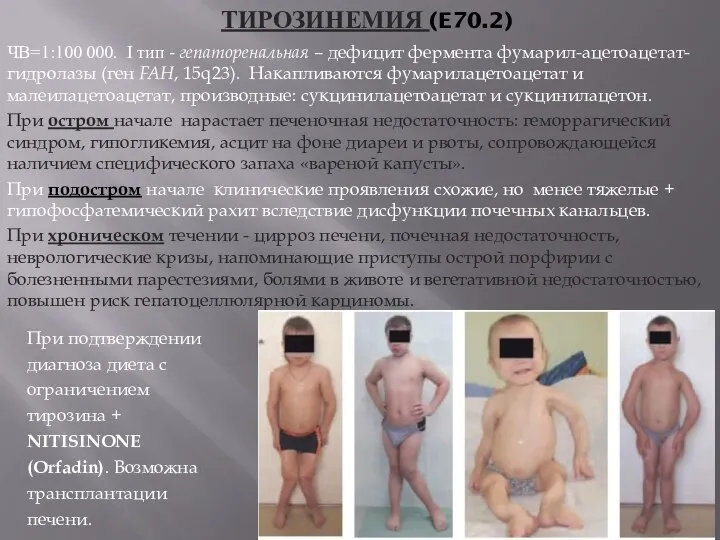
ТИРОЗИНЕМИЯ (E70.2)
ЧВ=1:100 000. I тип - гепаторенальная – дефицит фермента фумарил-ацетоацетат-гидролазы
ТИРОЗИНЕМИЯ (E70.2)
ЧВ=1:100 000. I тип - гепаторенальная – дефицит фермента фумарил-ацетоацетат-гидролазы

ТИРОЗИНЕМИЯ (E70.2)
2тип – глазокожная тирозинемия –дефицит фермента тирозинаминотрансферазы (ген TAT, 16q22.2).
ТИРОЗИНЕМИЯ (E70.2)
2тип – глазокожная тирозинемия –дефицит фермента тирозинаминотрансферазы (ген TAT, 16q22.2).

ТИРОЗИНЕМИЯ (E70.2)
3 тип
дефицит фермента 4гидроксифенил-пируватдиоксигеназы
(HPD, 12q24.31).
Характеризуется мягкой гипертирозинемией.
Описаны бессимптомные
ТИРОЗИНЕМИЯ (E70.2)
3 тип
дефицит фермента 4гидроксифенил-пируватдиоксигеназы
(HPD, 12q24.31).
Характеризуется мягкой гипертирозинемией.
Описаны бессимптомные

ЛЕЙЦИНОЗ (болезнь «кленового сиропа») (E71.0)
АР тип наследования, ЧВ=1:120 000.
ЭТИОЛОГИЯ: Мутации в
ЛЕЙЦИНОЗ (болезнь «кленового сиропа») (E71.0)
АР тип наследования, ЧВ=1:120 000.
ЭТИОЛОГИЯ: Мутации в

ЛЕЙЦИНОЗ (болезнь «кленового сиропа») (E71.0)
Клиника: 4 варианта течения (при всех будет
ЛЕЙЦИНОЗ (болезнь «кленового сиропа») (E71.0)
Клиника: 4 варианта течения (при всех будет

ГОМОЦИСТИНУРИЯ (E72.1)
Нарушение обмена аминокислоты метионина.
АР тип наследования, ген 21q22.1. ЧВ=1:200
ГОМОЦИСТИНУРИЯ (E72.1)
Нарушение обмена аминокислоты метионина.
АР тип наследования, ген 21q22.1. ЧВ=1:200

ГОМОЦИСТИНУРИЯ (E72.1)
Диагностика: БХ анализ крови и мочи (высокое содержание гомоцистина и
ГОМОЦИСТИНУРИЯ (E72.1)
Диагностика: БХ анализ крови и мочи (высокое содержание гомоцистина и
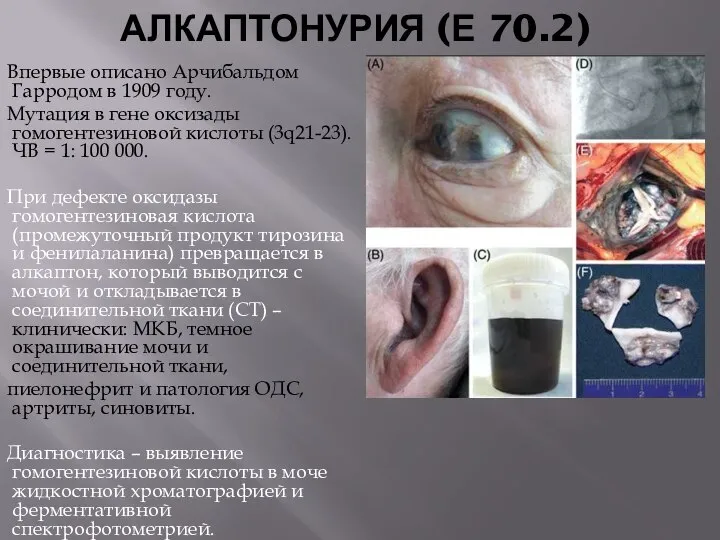
АЛКАПТОНУРИЯ (Е 70.2)
Впервые описано Арчибальдом Гарродом в 1909 году.
Мутация в гене
АЛКАПТОНУРИЯ (Е 70.2)
Впервые описано Арчибальдом Гарродом в 1909 году.
Мутация в гене
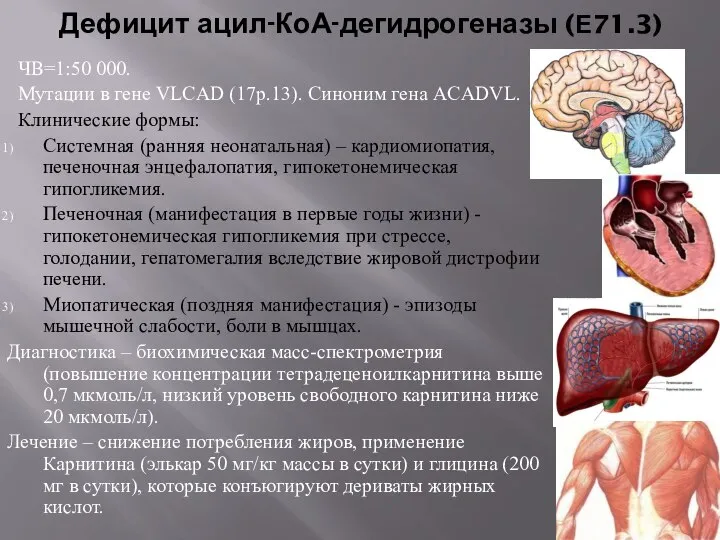
Дефицит ацил-КоА-дегидрогеназы (Е71.3)
ЧВ=1:50 000.
Мутации в гене VLCAD (17p.13). Синоним гена ACADVL.
Клинические
Дефицит ацил-КоА-дегидрогеназы (Е71.3)
ЧВ=1:50 000.
Мутации в гене VLCAD (17p.13). Синоним гена ACADVL.
Клинические

Болезнь Леша-Нихена (Е79.1)
ЧВ=1:300 000. (В 1964 г. – Майкл Лёш и
Болезнь Леша-Нихена (Е79.1)
ЧВ=1:300 000. (В 1964 г. – Майкл Лёш и
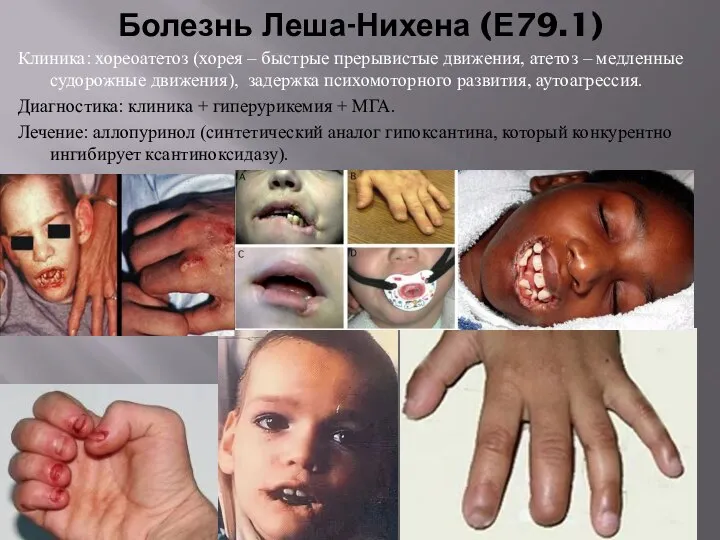
Болезнь Леша-Нихена (Е79.1)
Клиника: хореоатетоз (хорея – быстрые прерывистые движения, атетоз –
Болезнь Леша-Нихена (Е79.1)
Клиника: хореоатетоз (хорея – быстрые прерывистые движения, атетоз –
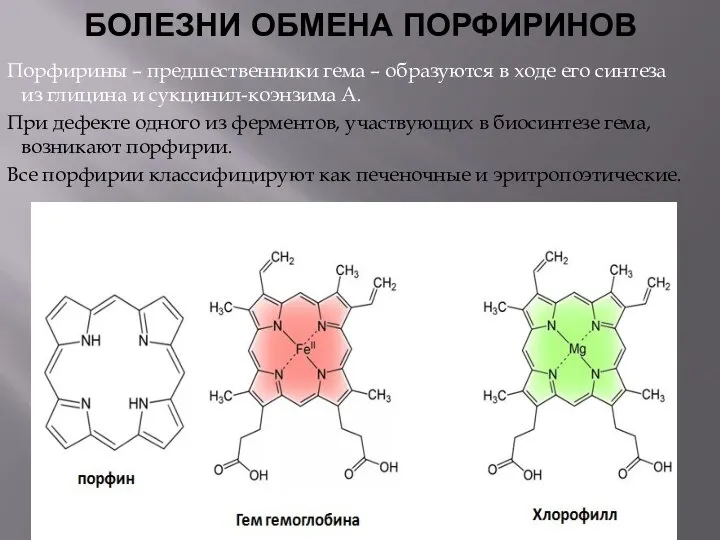
БОЛЕЗНИ ОБМЕНА ПОРФИРИНОВ
Порфирины – предшественники гема – образуются в ходе его
БОЛЕЗНИ ОБМЕНА ПОРФИРИНОВ
Порфирины – предшественники гема – образуются в ходе его

Острая перемежающаяся порфирия (Е80.2)
ЧВ=1:100 000 населения. АД тип наследования, неполная пенетрантность.
Мутация
Острая перемежающаяся порфирия (Е80.2)
ЧВ=1:100 000 населения. АД тип наследования, неполная пенетрантность.
Мутация

МУКОВИСЦЕДОЗ (E84.0)
Мутация в гене муковисцедозного трансмембранного регулятора проводимости, 7q31-32.
ЧВ=1:5000.
Ген экспрессируется
МУКОВИСЦЕДОЗ (E84.0)
Мутация в гене муковисцедозного трансмембранного регулятора проводимости, 7q31-32.
ЧВ=1:5000.
Ген экспрессируется

БОЛЕЗНИ ОБМЕНА ГОРМОНОВ
Строение гонана (циклопентан-пергидрофенантрена), основы строения стероидов
БОЛЕЗНИ ОБМЕНА ГОРМОНОВ
Строение гонана (циклопентан-пергидрофенантрена), основы строения стероидов
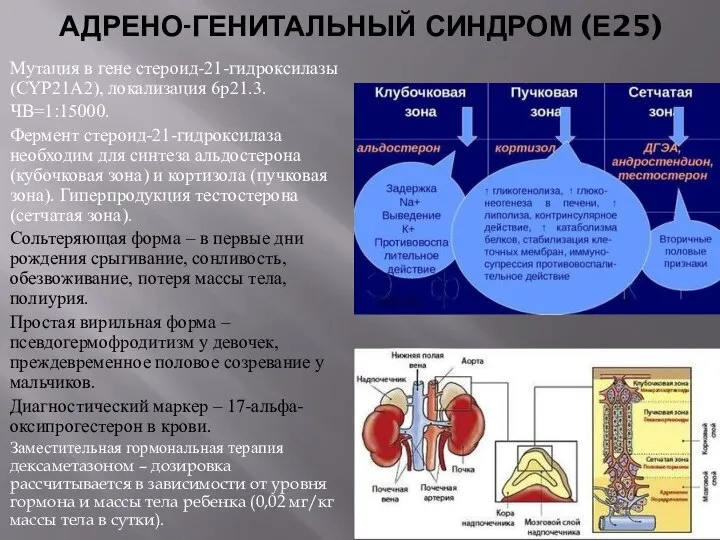
АДРЕНО-ГЕНИТАЛЬНЫЙ СИНДРОМ (Е25)
Мутация в гене стероид-21-гидроксилазы (CYP21А2), локализация 6р21.3.
ЧВ=1:15000.
Фермент стероид-21-гидроксилаза необходим
АДРЕНО-ГЕНИТАЛЬНЫЙ СИНДРОМ (Е25)
Мутация в гене стероид-21-гидроксилазы (CYP21А2), локализация 6р21.3.
ЧВ=1:15000.
Фермент стероид-21-гидроксилаза необходим
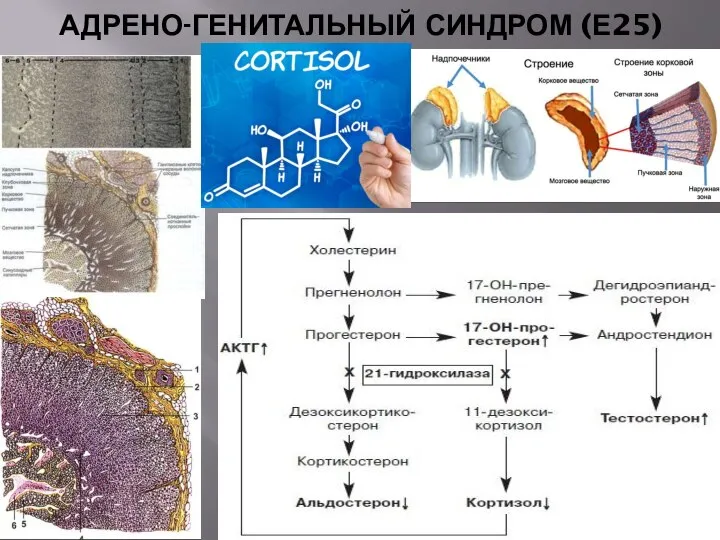
АДРЕНО-ГЕНИТАЛЬНЫЙ СИНДРОМ (Е25)
АДРЕНО-ГЕНИТАЛЬНЫЙ СИНДРОМ (Е25)

ВРОЖДЕННЫЙ ГИПОТИРЕОЗ (Е00)
Мутации в генах TITF1 (тиреоидный фактор транскрипции), локализация 14q13-21.
ЧВ=1:5000.
Крупный
ВРОЖДЕННЫЙ ГИПОТИРЕОЗ (Е00)
Мутации в генах TITF1 (тиреоидный фактор транскрипции), локализация 14q13-21.
ЧВ=1:5000.
Крупный
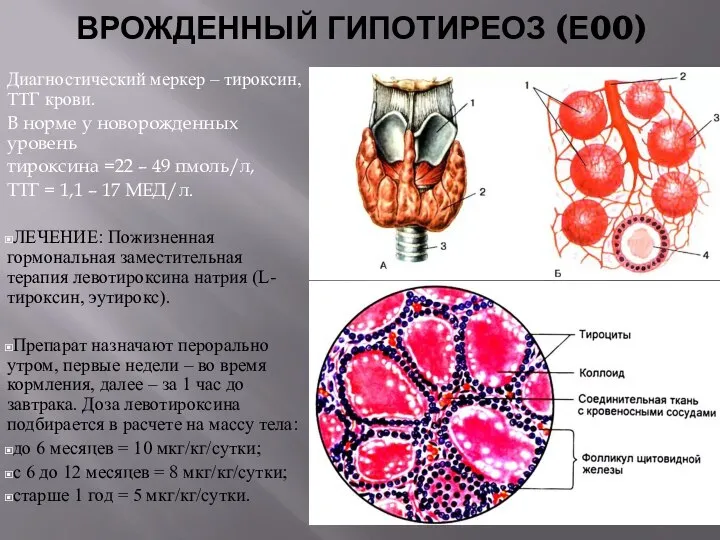
ВРОЖДЕННЫЙ ГИПОТИРЕОЗ (Е00)
Диагностический меркер – тироксин, ТТГ крови.
В норме у новорожденных
ВРОЖДЕННЫЙ ГИПОТИРЕОЗ (Е00)
Диагностический меркер – тироксин, ТТГ крови.
В норме у новорожденных
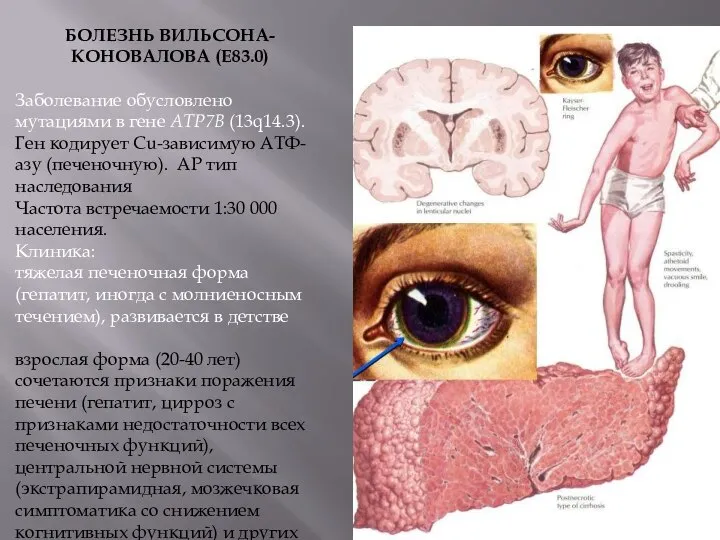
БОЛЕЗНЬ ВИЛЬСОНА-КОНОВАЛОВА (Е83.0)
Заболевание обусловлено мутациями в гене АТР7В (13q14.3).
Ген кодирует
БОЛЕЗНЬ ВИЛЬСОНА-КОНОВАЛОВА (Е83.0)
Заболевание обусловлено мутациями в гене АТР7В (13q14.3).
Ген кодирует

Патогномоничным симптомом заболевания является зеленовато-бурое кольцо отложений меди по периферии радужной
Патогномоничным симптомом заболевания является зеленовато-бурое кольцо отложений меди по периферии радужной

БОЛЕЗНЬ МЕНКЕСА (Е83.0)
Ген АТР7А (Xq21.1). Ген кодирует Cu-зависимую АТФ-азу (кишечную), в
БОЛЕЗНЬ МЕНКЕСА (Е83.0)
Ген АТР7А (Xq21.1). Ген кодирует Cu-зависимую АТФ-азу (кишечную), в
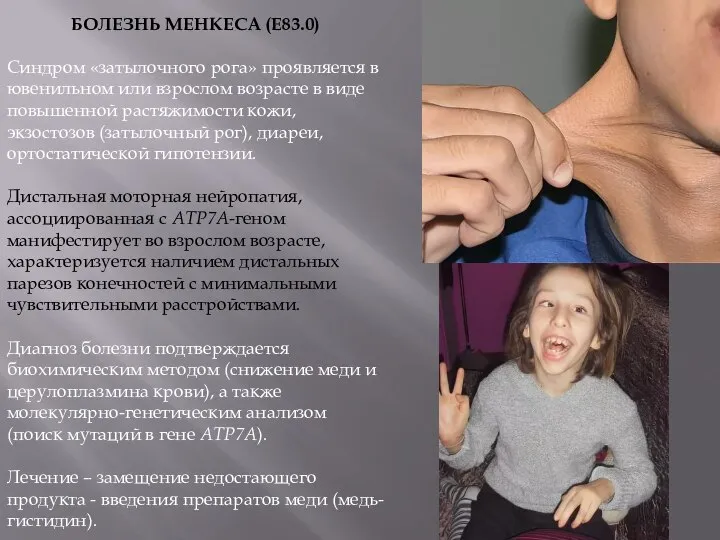
БОЛЕЗНЬ МЕНКЕСА (Е83.0)
Синдром «затылочного рога» проявляется в ювенильном или взрослом возрасте
БОЛЕЗНЬ МЕНКЕСА (Е83.0)
Синдром «затылочного рога» проявляется в ювенильном или взрослом возрасте

ГЕМОХРОМАТОЗ (Е83.1)
Накопление избытка Fe в органах, c их тяжелым поражением (цирроз,
ГЕМОХРОМАТОЗ (Е83.1)
Накопление избытка Fe в органах, c их тяжелым поражением (цирроз,
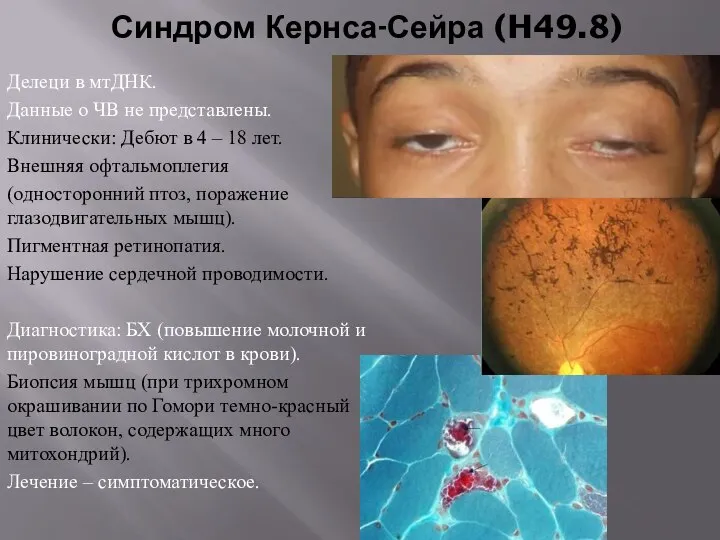
Синдром Кернса-Сейра (H49.8)
Делеци в мтДНК.
Данные о ЧВ не представлены.
Клинически: Дебют в
Синдром Кернса-Сейра (H49.8)
Делеци в мтДНК.
Данные о ЧВ не представлены.
Клинически: Дебют в

Синдром MELAS (G71.3)
Митохондриальная энцефалопатия, лактоацидоз и инсультоподобные эпизоды. ЧВ=1:10 000 человек.
Мутации
Синдром MELAS (G71.3)
Митохондриальная энцефалопатия, лактоацидоз и инсультоподобные эпизоды. ЧВ=1:10 000 человек.
Мутации

Синдром MERRF (G71.3)
Миоклоническая эпилепсия с рваными мышечными волокнами.
Мутации в мтДНК: MTTK,
Синдром MERRF (G71.3)
Миоклоническая эпилепсия с рваными мышечными волокнами.
Мутации в мтДНК: MTTK,

Мукополисахаридозы
Мукополисахаридозы – группа заболеваний, обусловленных генетическим дефектом ферментного расщепления углеводной части
Мукополисахаридозы
Мукополисахаридозы – группа заболеваний, обусловленных генетическим дефектом ферментного расщепления углеводной части

МУКОПОЛИСАХАРИДОЗЫ
МПС
МУКОПОЛИСАХАРИДОЗЫ
МПС

МПС I типа – синдром Гурлера (Е76.0)
ЧВ= 1:100 000 населения.
МПС I типа – синдром Гурлера (Е76.0)
ЧВ= 1:100 000 населения.
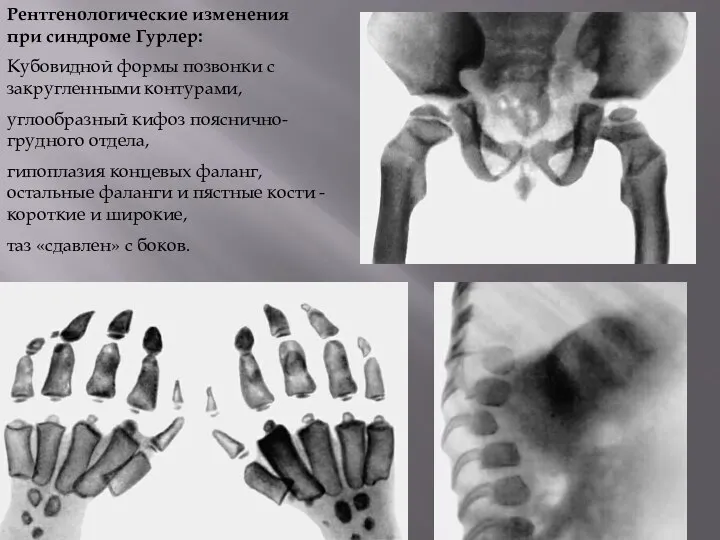
Рентгенологические изменения при синдроме Гурлер:
Кубовидной формы позвонки с закругленными контурами,
Рентгенологические изменения при синдроме Гурлер:
Кубовидной формы позвонки с закругленными контурами,
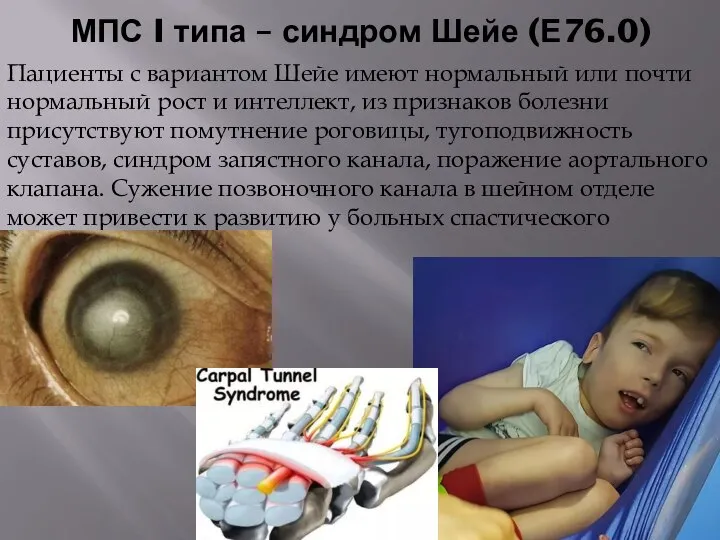
МПС I типа – синдром Шейе (Е76.0)
Пациенты с вариантом Шейе
МПС I типа – синдром Шейе (Е76.0)
Пациенты с вариантом Шейе
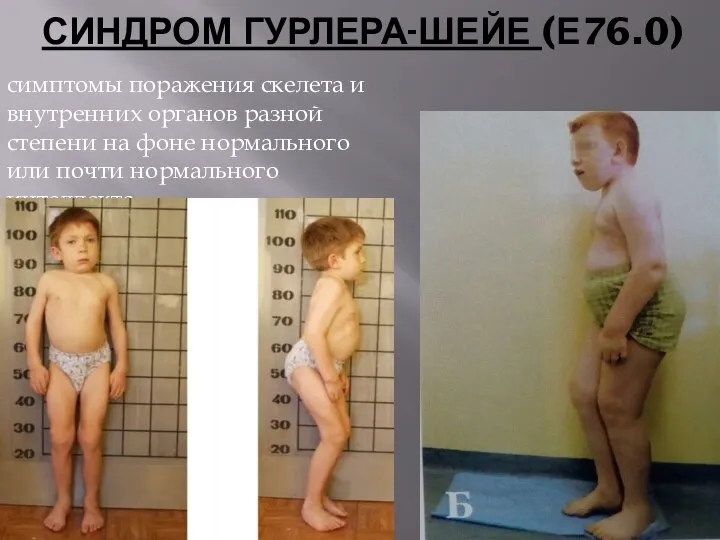
СИНДРОМ ГУРЛЕРА-ШЕЙЕ (Е76.0)
симптомы поражения скелета и внутренних органов разной степени на
СИНДРОМ ГУРЛЕРА-ШЕЙЕ (Е76.0)
симптомы поражения скелета и внутренних органов разной степени на

МПС I типа
Диагностика:
биохимический метод (повышение в моче уровня дерматансульфатов и гепарансульфатов
МПС I типа
Диагностика:
биохимический метод (повышение в моче уровня дерматансульфатов и гепарансульфатов

Синдром Хантера (Е76.1)
ЧВ= 1:132 000 населения. Ген IDS, Xq28. ИДУРОНАТ-L-СУЛЬФАТАЗА
КП: когнитивные
Синдром Хантера (Е76.1)
ЧВ= 1:132 000 населения. Ген IDS, Xq28. ИДУРОНАТ-L-СУЛЬФАТАЗА
КП: когнитивные

Синдром Санфилиппо (Е76.2)
Тип А (1:100 000, SGSH, 17q25.3, СУЛЬФОГИДРОЛАЗА N-СУЛЬФОГЛЮКОЗАМИНА),
тип B
Синдром Санфилиппо (Е76.2)
Тип А (1:100 000, SGSH, 17q25.3, СУЛЬФОГИДРОЛАЗА N-СУЛЬФОГЛЮКОЗАМИНА),
тип B
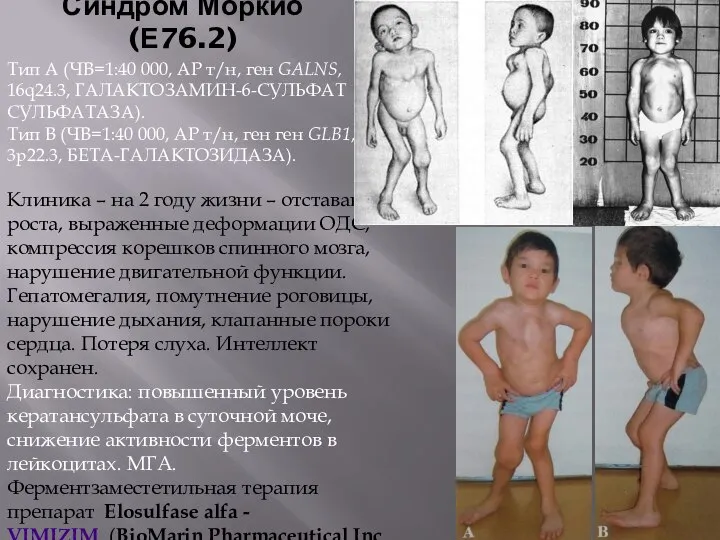
Синдром Моркио (Е76.2)
Тип А (ЧВ=1:40 000, АР т/н, ген GALNS, 16q24.3,
Синдром Моркио (Е76.2)
Тип А (ЧВ=1:40 000, АР т/н, ген GALNS, 16q24.3,

СИНДРОМ МАРОТО-ЛАМИ (Е76.2)
ЧВ=1:215 000. АР т/н.
Ген ARSB, 5q14.1.
АРИЛСУЛЬФАТАЗА-В.
Накопление в организме
СИНДРОМ МАРОТО-ЛАМИ (Е76.2)
ЧВ=1:215 000. АР т/н.
Ген ARSB, 5q14.1.
АРИЛСУЛЬФАТАЗА-В.
Накопление в организме
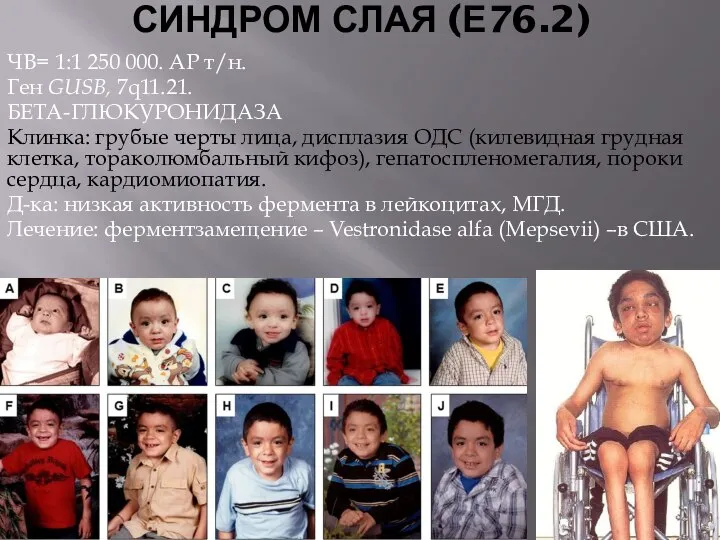
СИНДРОМ СЛАЯ (Е76.2)
ЧВ= 1:1 250 000. АР т/н.
Ген GUSB, 7q11.21.
БЕТА-ГЛЮКУРОНИДАЗА
Клинка:
СИНДРОМ СЛАЯ (Е76.2)
ЧВ= 1:1 250 000. АР т/н.
Ген GUSB, 7q11.21.
БЕТА-ГЛЮКУРОНИДАЗА
Клинка:
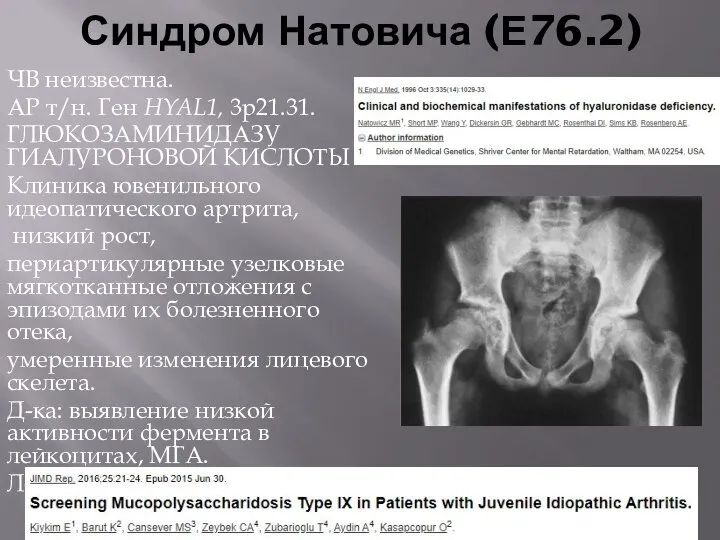
Синдром Натовича (Е76.2)
ЧВ неизвестна.
АР т/н. Ген HYAL1, 3p21.31.
ГЛЮКОЗАМИНИДАЗУ ГИАЛУРОНОВОЙ
Синдром Натовича (Е76.2)
ЧВ неизвестна.
АР т/н. Ген HYAL1, 3p21.31.
ГЛЮКОЗАМИНИДАЗУ ГИАЛУРОНОВОЙ

СФИНГОЛИПИДОЗЫ
При сфинголипидозах – внутриклеточное накопление сфинголипидов в головном и костном мозге,
СФИНГОЛИПИДОЗЫ
При сфинголипидозах – внутриклеточное накопление сфинголипидов в головном и костном мозге,

Niemann-Pick diseases (Е75.2)
ЧВ типа А и В = 1:40 000, типа
Niemann-Pick diseases (Е75.2)
ЧВ типа А и В = 1:40 000, типа

Fabry disease (Е75.2)
ЧВ=1:40 000. Ген GLA, Xq22.1 (альфа-галактозидаза А).
Накопление глоботриаозилцерамида.
Клиника: грубые
Fabry disease (Е75.2)
ЧВ=1:40 000. Ген GLA, Xq22.1 (альфа-галактозидаза А).
Накопление глоботриаозилцерамида.
Клиника: грубые

GM1-ГАНГЛИОЗИДОЗ (БОЛЕЗНЬ ТЕЯ-САКСА) (E75.0)
АР тип наследования.
Ген HEXA, 15q23, альфа субъединица гексаминидазы.
ЧВ=1:320000,
GM1-ГАНГЛИОЗИДОЗ (БОЛЕЗНЬ ТЕЯ-САКСА) (E75.0)
АР тип наследования.
Ген HEXA, 15q23, альфа субъединица гексаминидазы.
ЧВ=1:320000,

Gaucher’s disease(E75.2)
Ген GBA, 1q22. БЕТА-ГЛЮКОЦЕРЕБРОЗИДАЗА.
Накопление глюкоцереброзида – клетки Гоше.
Тип I
Gaucher’s disease(E75.2)
Ген GBA, 1q22. БЕТА-ГЛЮКОЦЕРЕБРОЗИДАЗА.
Накопление глюкоцереброзида – клетки Гоше.
Тип I

БГ тип 1 до и после ферментзаместительной терапии
Девочка 11 мес, БГ
БГ тип 1 до и после ферментзаместительной терапии
Девочка 11 мес, БГ

БОЛЕЗНЬ КРАББЕ (E75.2)
ЧВ=1:100 000, ген GALC, 14q31.
ГАЛАКТОЦЕРЕБРОЗИДАЗА.
Накопление в ЦНС галактозилерамидов.
Дебют
БОЛЕЗНЬ КРАББЕ (E75.2)
ЧВ=1:100 000, ген GALC, 14q31.
ГАЛАКТОЦЕРЕБРОЗИДАЗА.
Накопление в ЦНС галактозилерамидов.
Дебют

ПЕРОКСИСОМНЫЕ БОЛЕЗНИ
Пероксисома - обязательная органелла эукариотической клетки, ограниченная мембраной, содержащая от
ПЕРОКСИСОМНЫЕ БОЛЕЗНИ
Пероксисома - обязательная органелла эукариотической клетки, ограниченная мембраной, содержащая от

1. Пероксисомы получили такое название благодаря тому, что обычно в их
1. Пероксисомы получили такое название благодаря тому, что обычно в их

В пероксисомах происходит окисление длинноцепочечных жирных кислот с образованием ацил-CoA, который
В пероксисомах происходит окисление длинноцепочечных жирных кислот с образованием ацил-CoA, который

АДРЕНОЛЕЙКОДИСТРОФИЯ (Е71.3)
ЧВ=1:20 000. Ген ABCD1, Xq28. Кодирует транспортер жирных кислот с
АДРЕНОЛЕЙКОДИСТРОФИЯ (Е71.3)
ЧВ=1:20 000. Ген ABCD1, Xq28. Кодирует транспортер жирных кислот с

Refsum disease (G60.1)
Ген PHYH, 6q22 – ФИТАНОИЛ-КОА-ГИДРОКСИЛАЗА.
ЧВ=1:25 000.
Накопление фитановой кислоты в
Refsum disease (G60.1)
Ген PHYH, 6q22 – ФИТАНОИЛ-КОА-ГИДРОКСИЛАЗА.
ЧВ=1:25 000.
Накопление фитановой кислоты в

СИНДРОМ ЦЕЛЬВЕГЕРА (Q87.8)
АР т/н. ЧВ=1:50 000.
Гены PEX1, 2, 3, 5, 6,
СИНДРОМ ЦЕЛЬВЕГЕРА (Q87.8)
АР т/н. ЧВ=1:50 000.
Гены PEX1, 2, 3, 5, 6,

Синдром Цельвегера
Скафоцефалия (преждевременное
зарастание швов черепа).
Гипоплазия носа.
Выпуклый куполообразный лоб.
Низкопосаженные уши.
Ретрогнатия (сдвиг
Синдром Цельвегера
Скафоцефалия (преждевременное
зарастание швов черепа).
Гипоплазия носа.
Выпуклый куполообразный лоб.
Низкопосаженные уши.
Ретрогнатия (сдвиг

Принципы патогенетического лечения НБОВ:
Устранение поступления субстрата с пищей.
Коррекция выведения продукта (болезнь
Принципы патогенетического лечения НБОВ:
Устранение поступления субстрата с пищей.
Коррекция выведения продукта (болезнь

Гемофилии D66
Гемофилия А. Ген VIII п.ф.с., Xq28, ЧВ=1:10 000.
Гемофилия В. Ген
Гемофилии D66
Гемофилия А. Ген VIII п.ф.с., Xq28, ЧВ=1:10 000.
Гемофилия В. Ген

НАСЛЕДСТВЕННЫЕ ДИСЛИПИДЕМИИ
1 тип – гиперхиломикронемия (ген липопротеинлипазы- 8p22, ген апоС2 –
НАСЛЕДСТВЕННЫЕ ДИСЛИПИДЕМИИ
1 тип – гиперхиломикронемия (ген липопротеинлипазы- 8p22, ген апоС2 –

Наследственные иммунодефициты.
Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1
Наследственные иммунодефициты. Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1

ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов
ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов

АДА-дефицит составляет около 15% всех случаев ТКИД и около трети –
АДА-дефицит составляет около 15% всех случаев ТКИД и около трети –

В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что
В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что

1) по типу клеток-мишеней:
соматическая
фетальная
2) по цели воздействия:
позитивная
1) по типу клеток-мишеней:
соматическая
фетальная
2) по цели воздействия:
позитивная

Вирусные векторы:
ретровирусы
аденовирусы
аденоассоциированный вирус
герпесвирусы
лентивирусы
и др.
- достоинства:
Вирусные векторы:
ретровирусы
аденовирусы
аденоассоциированный вирус
герпесвирусы
лентивирусы
и др.
- достоинства:

Аденовирусы
Преимущества:
способны инфицировать неделящиеся клетки
большая клонирующая емкость (в настоящее время
Аденовирусы
Преимущества:
способны инфицировать неделящиеся клетки
большая клонирующая емкость (в настоящее время
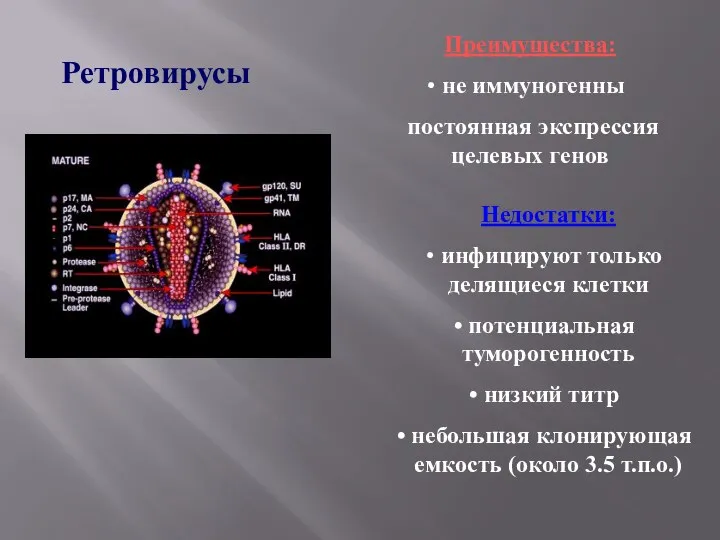
Ретровирусы
Преимущества:
не иммуногенны
постоянная экспрессия целевых генов
Недостатки:
инфицируют только делящиеся клетки
Ретровирусы
Преимущества:
не иммуногенны
постоянная экспрессия целевых генов
Недостатки:
инфицируют только делящиеся клетки

Невирусные системы
прямая инъекция
рецепторо-опосредованный эндоцитоз
генное ружье
липофекция
электропорация
полимерные
Невирусные системы
прямая инъекция
рецепторо-опосредованный эндоцитоз
генное ружье
липофекция
электропорация
полимерные

Плазмидные векторы
- достоинства:
отсутствие токсичности и мутагенности
практически неограниченная емкость вектора
Плазмидные векторы
- достоинства:
отсутствие токсичности и мутагенности
практически неограниченная емкость вектора

Клинические испытания генотерапевтических препаратов.
I фаза. Оценка токсичности генной конструкции.
II
Клинические испытания генотерапевтических препаратов.
I фаза. Оценка токсичности генной конструкции.
II

ПРИМЕРЫ:
муковисцидоз (кистозный фиброз поджелудочной железы) - перенос гена МТР (муковисцидозный
ПРИМЕРЫ:
муковисцидоз (кистозный фиброз поджелудочной железы) - перенос гена МТР (муковисцидозный


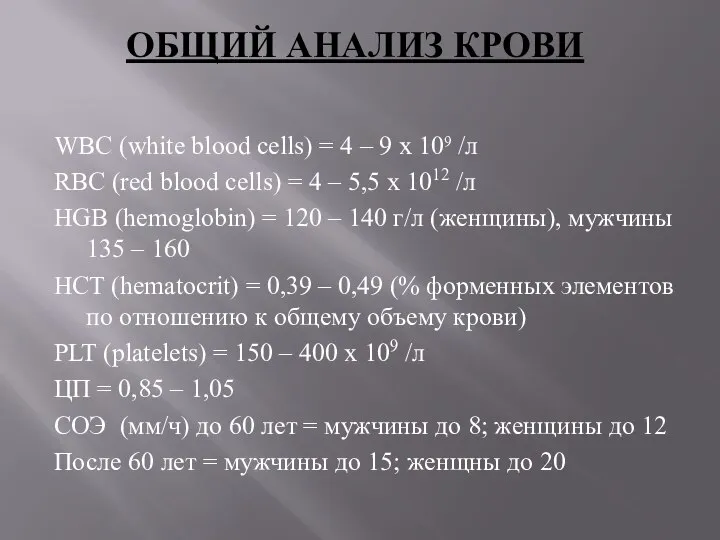
WBC (white blood cells) = 4 – 9 x 109 /л
RBC
WBC (white blood cells) = 4 – 9 x 109 /л
RBC

По степени тяжести: легкой ( Hb >90 г/л), средней (70 –
По степени тяжести: легкой ( Hb >90 г/л), средней (70 –

Гемолитическая анемия
Гемолитическая анемия возникает при преобладании процесса разрушения эритроцитов над их
Гемолитическая анемия
Гемолитическая анемия возникает при преобладании процесса разрушения эритроцитов над их

Апластическая анемия Фалькони .
Гипоплатическая анемия Ерлиха.
Анемия Даймонда-Блекфена.
Синдром Швахмана-Даймонда.
Наследственные апластические анемии
Апластическая анемия Фалькони .
Гипоплатическая анемия Ерлиха.
Анемия Даймонда-Блекфена.
Синдром Швахмана-Даймонда.
Наследственные апластические анемии

МЕМБРАНОПАТИИ
сфероцитоз Минковского-Шоффара
наследственный элиптоцитоз
наследственный пиропойкилоцитоз
наследственный стоматоцитоз
наследственный акантоцитоз
наследственный эхиноцитоз
МЕМБРАНОПАТИИ
сфероцитоз Минковского-Шоффара
наследственный элиптоцитоз
наследственный пиропойкилоцитоз
наследственный стоматоцитоз
наследственный акантоцитоз
наследственный эхиноцитоз

АД тип наследования. ЧВ= 1 : 5000 населения.
Около 25% случаев спорадические.
Мутации
АД тип наследования. ЧВ= 1 : 5000 населения.
Около 25% случаев спорадические.
Мутации

манифестация в раннем детском возрасте
ДИАГНОСТИКА:
-наследственный анамнез.
-осмотр (башенный череп, готическое небо, изменения
манифестация в раннем детском возрасте
ДИАГНОСТИКА:
-наследственный анамнез.
-осмотр (башенный череп, готическое небо, изменения


Наследственный сфероцитоз
Лечение:
Бессимптомные формы
Лечения не требуется
УЗИ контроль состояния желчных путей
Легкая и
Наследственный сфероцитоз
Лечение:
Бессимптомные формы
Лечения не требуется
УЗИ контроль состояния желчных путей
Легкая и

Ферментопатии
Недостаточность эритроцитарных ферментов:
глюкозо-6-фосфатдегидрогеназа
пируваткиназа
глюкозофосфат изомераза
Ферментопатии
Недостаточность эритроцитарных ферментов:
глюкозо-6-фосфатдегидрогеназа
пируваткиназа
глюкозофосфат изомераза

ген - на Х-хромосоме (Xq28) – наследование Х-сцепленное, болеют мальчики; редко
ген - на Х-хромосоме (Xq28) – наследование Х-сцепленное, болеют мальчики; редко

острая гемолитическая анемия
развивается спустя несколько часов или дней от начала приема
острая гемолитическая анемия
развивается спустя несколько часов или дней от начала приема

Дефицит глюкозо-6-фосфат дегидрогеназы (Г-6-ФД)
Диагностика:
Признаки внутрисосудистого гемолиза – увеличение непрямого билирубина, ЛДГ,
Дефицит глюкозо-6-фосфат дегидрогеназы (Г-6-ФД)
Диагностика:
Признаки внутрисосудистого гемолиза – увеличение непрямого билирубина, ЛДГ,

Серповидно-клеточная анемия
АР тип наследования.
В норме гемоглобин человека состоит из 2
Серповидно-клеточная анемия
АР тип наследования.
В норме гемоглобин человека состоит из 2

ГЕМОГЛОБИНОПАТИИ
Серповидноклеточная анемия. У таких больных вместо гемоглобина А синтезируется гемоглобин S.
ГЕМОГЛОБИНОПАТИИ
Серповидноклеточная анемия. У таких больных вместо гемоглобина А синтезируется гемоглобин S.

Серповидноклеточная анемия. Клинические проявления.
Конституциональные проявления – отставание роста и развития, полового
Серповидноклеточная анемия. Клинические проявления.
Конституциональные проявления – отставание роста и развития, полового

Серповидноклеточная анемия. Клинические проявления.
Апластические кризы. Пациенты с СКА подвержены инфекциям и
Серповидноклеточная анемия. Клинические проявления.
Апластические кризы. Пациенты с СКА подвержены инфекциям и
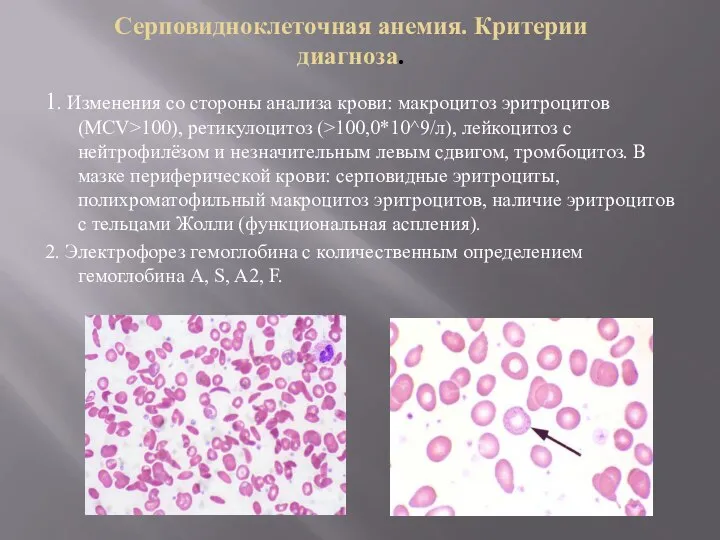
Серповидноклеточная анемия. Критерии диагноза.
1. Изменения со стороны анализа крови: макроцитоз эритроцитов
Серповидноклеточная анемия. Критерии диагноза.
1. Изменения со стороны анализа крови: макроцитоз эритроцитов

Серповидно-клеточная анемия
Терапия
Инфузионная терапия, анальгетики
Трансфузионная терапия – только по показаниям (с осторожностью!!!,
Серповидно-клеточная анемия
Терапия
Инфузионная терапия, анальгетики
Трансфузионная терапия – только по показаниям (с осторожностью!!!,

ТАЛАССЕМИЯ
Это группа аутосомно – рецессивных заболеваний крови, характеризующихся снижением синтеза одного
ТАЛАССЕМИЯ
Это группа аутосомно – рецессивных заболеваний крови, характеризующихся снижением синтеза одного

Молекулярные основы талассемий.
Синтез a – цепей глобина кодируется диплоидным набором генов
Молекулярные основы талассемий.
Синтез a – цепей глобина кодируется диплоидным набором генов

Молекулярные основы талассемий.
Малая талассемия (В+/В) протекает, как правило, без клинических проявлений,
Молекулярные основы талассемий.
Малая талассемия (В+/В) протекает, как правило, без клинических проявлений,

Талассемии. Клинические проявления.
1) Анемия хронического течения, вызывающая замедление роста, полового развития,
Талассемии. Клинические проявления.
1) Анемия хронического течения, вызывающая замедление роста, полового развития,

Талассемии. Критерии диагноза.
А. В анализе крови анемия различной степени, микроцитоз эритроцитов
Талассемии. Критерии диагноза.
А. В анализе крови анемия различной степени, микроцитоз эритроцитов

Талассемии. Лечение.
При тяжёлых формах:
1) Регулярные трансфузии эритроцитарной массы (1 – 3
Талассемии. Лечение.
При тяжёлых формах:
1) Регулярные трансфузии эритроцитарной массы (1 – 3
 Середня дорослість (Е. Еріксон)
Середня дорослість (Е. Еріксон) Профилактика и организация медицинской помощи при злокачественных новообразованиях
Профилактика и организация медицинской помощи при злокачественных новообразованиях Клиническая фармакология пожилых и беременных
Клиническая фармакология пожилых и беременных Патогенез лейкозов
Патогенез лейкозов Дерматомиозит
Дерматомиозит Вирусные гепатиты
Вирусные гепатиты Заболевания глаз
Заболевания глаз Первая помощь при кровотечении
Первая помощь при кровотечении Нейроэндокринные опухоли
Нейроэндокринные опухоли Аллергия (гиперчувствительность)
Аллергия (гиперчувствительность) Возрастная периодизация Д.Б. Эльконина
Возрастная периодизация Д.Б. Эльконина Robert Koch
Robert Koch Демографические показатели в оценке состояния общественного здоровья
Демографические показатели в оценке состояния общественного здоровья Общая характеристика зубных протезов
Общая характеристика зубных протезов Первичная профилактика патологического старения
Первичная профилактика патологического старения Средства, влияющие на функции органов дыхания
Средства, влияющие на функции органов дыхания Эритропоэздің нейро-гуморальдық реттелуі
Эритропоэздің нейро-гуморальдық реттелуі Прояви емоцій. Психічні стани, пов’язані з емоціями
Прояви емоцій. Психічні стани, пов’язані з емоціями Методы анализа белка в биологическом материале
Методы анализа белка в биологическом материале Как человек реагирует на явления в жизни и искусстве
Как человек реагирует на явления в жизни и искусстве Ультразвуковое исследование поджелудочной железы
Ультразвуковое исследование поджелудочной железы Потенция
Потенция Влияние ингибиторов гистондеацетилаз на формирование памяти
Влияние ингибиторов гистондеацетилаз на формирование памяти Анатомия наружного и среднего уха
Анатомия наружного и среднего уха Способы, с помощью которых возможно избежать последствия хронического недосыпания
Способы, с помощью которых возможно избежать последствия хронического недосыпания Здоровое питание и мой ребенок
Здоровое питание и мой ребенок Синдром анемии в обще-врачебной практике
Синдром анемии в обще-врачебной практике Способы и методика введения лекарственных средств детям
Способы и методика введения лекарственных средств детям