- Острые лейкозы

Содержание
- 2. Онкогематологические заболевания (гемобластозы) – злокачественные новообразования, морфологическим субстратом которых являются неопластически трансформированные кроветворные клетки . Гемобластозы,
- 3. Максимов А.А. в 1908г впервые высказал представление о наличии единой клетки-предшественницы для всех ростков кроветворения –
- 5. Клеточный состав костного мозга % Клеточный состав костного мозга – 50 – 250 кл в 1
- 6. Распространенность гемобластозов Суммарная частота гемобластозов: 25–30 случаев на 100 000 населения в год Наиболее распространены неходжкинские
- 7. Распределение гемобластозов Все онкогематологические заболевания, за исключением острого лимфобластного лейкоза, лимфобластной лимфомы и лимфомы Беркитта, значительно
- 8. Этиологические (предрасполагающие) факторы гемобластозов В основе - первичная мутации кроветворной клетки-предшественницы под действием мутагенов – хромосомные
- 9. Патогенез онкогематологических заболеваний Гемобластозы возникают вследствие мутации клетки-предшественницы, приводящей к клональной неопластической пролиферации злокачественно трансформированных кроветворных
- 10. Патогенез онкогематологических заболеваний (продолжение) При лабораторных признаках лейкозов в организме больного имеется около 1011 бластных клеток,
- 11. Патогенез онкогематологических заболеваний (окончание) Для гемобластозов характерна опухолевая прогрессия: по мере развития заболевания появляются клоны более
- 12. Острые лейкозы – группа клональных заболеваний системы крови, первично поражающих костный мозг, морфологическим субстратом которых являются
- 13. Все острые лейкозы возникают из одной мутировавшей клетки, которая может относиться как к очень ранним, так
- 14. Острые лейкозы – отдельная группа онкогематологических заболеваний, которые никогда не трансформируются в хронические лейкозы. Острый лейкоз
- 15. Выделяют два вида заболевания, которые различаются по морфологии, течению, характеру проводимой химиотерапии и результатам лечения: 1)
- 16. FAB - классификация острых лейкозов В 1976 году группой экспертов Франции, США и Великобритании (FAB) была
- 17. Для верификации варианта острого лейкоза и его варианта требуются цитохимические, результаты которых имеют прогностическое значение и
- 18. Иммунофенотипирование Внедрение в практику моноклональных антител позволило идентифицировать специфические рецепторы, антигены и другие молекулы (маркеры) на
- 19. Цитогенетическое исследование У 90% больных ОЛ находят генетические поломки (транслокации, делеции, инверсии, гиперплоидию…). При ОЛЛ транслокация
- 20. Основные клинические синдромы при ОЛ Синдром лейкемической пролиферации: лимфоаденопатия, гепатоспленомегалия, расширение средостения; инфильтрация десен и яичек,
- 21. Синдром лейкемической - пролиферации при острых лейкозах
- 22. Проявления геморрагического cиндрома при ОЛ, в том числе в головной мозг, ЖК-тракт Инфекционные осложнения При ОЛ
- 23. Нейролейкоз Инфильтрация мозговых оболочек, приведшая к поражению лицевого нерва и одностороннему параличу мимических мышц Отек и
- 24. Варианты начала острого лейкоза - примерно у 50% больных начинается остро с клиники ангины, пневмонии, инфекции
- 25. Особенности течения некоторых форм ОМЛ О недифференцированный лейкоз (М0): частота его уменьшается в связи появляющимися дополнительными
- 26. О. монобластный лейкоз (М5) – 10%, клиника схожа с М4, экстрамедуллярные очаги встречаются еще чаще- до
- 27. О. Лимфобластный лейкоз – 10-15% (до 25%) взрослых больных ОЛ, (85% детей). Характено частое развитие синдрома
- 28. Клинический анализ крови Количество лейкоцитов периферической крови чаще остается на сублейкемическом уровне и не превышает 20–30х109/л
- 29. Пример клинического анализа крови больного ОЛ Гемоглобин – 79г/л; эритроциты – 2,8×10.12; цветовой показатель- 0,9; лейкоциты
- 30. В типичных случаях между бластами и зрелыми гранулоцитами отсутствуют промежуточные формы клеток нейтрофильного ряда (“лейкемический провал”
- 31. Клинический анализ крови- ( морфологическое исследование) При подсчете лейкоцитарной формулы у 90% больных выявляются бластные клетки,
- 32. Проведение костномозговой пункции обязательно!!! Иглы для проведения пункции: Игла Кассирского Современные иглы
- 33. Техника проведения стернальной пункции (трепанобиопсии) Область грудины Область подвздошной кости
- 34. Миелограмма- обязательна ! Количество миелокариоцитов обычно повышено, мегакариоциты отсутствуют или их количество снижено, сужение нормальных ростков
- 35. Для проведения морфологического исследования используются: 1 Клетки периферической крови 2 Мазки-отпечатки лимфоузла (опухоли) на предметном стекле
- 36. Существует правило: необходимо в течение всей жизни больного ОЛ хранить пунктаты костного мозга и мазки периферической
- 37. Стадии острого лейкоза I. Первая атака (первый острый период) – время от первых проявлений заболевания до
- 38. Стадии острого лейкоза (окончание) III. Рецидив – появление клинических и гематологических признаков ОЛ после полной ремиссии
- 39. Дифференциальный диагноз ОЛ 1 - заболевания, не являющимися гемобластозами: инфекционный мононуклеоз, Апластическая анемия, В12-дефицитная анемия. 2
- 40. Прогностические факторы при острых миелоидных лейкозах: 1) Значимые: - возраст более 60 лет – неблагоприятный прогноз;
- 41. Прогностически неблагоприятные факторы при остром лимфобластном лейкозе: - возраст более 35 лет; - лейкоцитоз более 300
- 42. Лечение острого лейкоза 1 исторический этап – 19 – начало 20 века – определение нозологической формы
- 43. Принципы химиотерапии острых лейкозов 1. Лечение необходимо начинать сразу же после установления диагноза, ( не только
- 44. 3. Выбор схемы лечения определяется морфологическим, цитохимическим и иммунофенотипическим, цитогенетическим вариантом острого лейкоза. 4. В большинстве
- 45. Основные методы лечения ОЛ 1 Цитостатическая терапия – уничтожение лейкозного клона; - восстановление нормального кроветворения 2
- 46. максимальная эрадикация лейкозного клона эрадикация минимальной резидуальной болезни, профилактика рецидива – 2-3 курса ДАТ: -цитозар 100мг/м2
- 47. максимальная эрадикация лейкозного клона эрадикация минимальной резидуальной болезни, профилактика рецидива – 2-3 курса CHOP : циклофосфан
- 48. Сопроводительная терапия лейкозов: 1 – Дезинтоксикационная терапия (физиологический раствор, полиглюкин, форсированный диурез и др.) 2 –
- 49. Осложнения острых лейкозов 1. Инфекционные осложнения (у 70% больных определяют летальные исходы). Для их профилактики за
- 50. 2 Геморрагический синдром - выраженная тромбоцитопения – у 1\2 больных - летальность от ГС – 5-10%
- 51. 3 Анемический синдром - анемия встречается у всех больных ОЛ, - основной метод лечения – переливание
- 52. 4 Энтеропатия Состояние, характеризующееся поражением слизистой ЖКТ, возникающее на фоне миелотоксического агранулоцитоза и проявляющееся болями в
- 53. 6 Нейролейкоз - частота при ОЛЛ – 30-50%; при ОНЛЛ- 5%. Клинические варианты: а) менингеальные симптомы
- 54. Современная химиотерапия позволяет получить полные ремиссии у 50–95% взрослых больных В большинстве случаев через 3-4 года
- 55. Лекция для 5 курса 4 факультета ФПВ на тему: « Хронические лейкозы »
- 56. Хронические лейкозы – гетерогенная группа опухолевых заболеваний крови, морфологическим субстратом которых является неопластически трансформированная клетка костного
- 57. Хронический лимфолейкоз (хронический лимфоцитарный лейкоз - клональное лимфопролиферативное заболевание, первично локализующееся в костном мозге, субстратом которого
- 59. Гемопоэз
- 60. Эпидемиология Составляет более 30% всех лейкозов в Северной Америке и Европе Ежегодная заболеваемость в этих регионах
- 61. Этиология ХЛЛ Для ХЛЛ не доказана этиологическая роль ионизирующего облучения. Для ХЛЛ животных доказана этиологическая роль
- 62. ХЛЛ СД5 Клетка-предшественница лимфопоэза Костный мозг СД19 СД23,24 Патогенез ХЛЛ
- 63. Патогенетические особенности ХЛЛ: - пролиферация клона трансформированных «зрелых» лимфоцитов; в иммунофенотипе - CD19, СД23, СД24, СД5+;
- 64. Клиническая картина В 70% случаев заболевание начинается постепенно и характеризуется лишь абсолютным лимфоцитозом в периферической крови
- 65. Синдром лимфатической пролиферации при ХЛЛ
- 66. 2 Симптомы опухолевой интоксикации: истощение, боли в костях, повышение температуры тела. 3 Аутоиммунные осложнения: гемолитическая анемия
- 67. Лабораторные данные Лейкоцитоз с абсолютным (более 5 × 109/л) и относительным лимфоцитозом; Морфологически лимфоциты не отличаются
- 68. Нормальные показатели клинического анализа крови
- 69. Клинический анализ крови при ХЛЛ
- 70. Миелограмма 1) При ХЛЛ; 2) нормальная
- 72. Критерии диагноза Абсолютный лимфоцитоз более 5 (10) 109/л Нормальная морфология лимфоцитов при световой микроскопии Более 30%
- 73. Классификация Rai
- 74. Критерии для начала лечения (Рано начатое лечение не увеличивает продолжительность жизни больных !!!) Симптомы интоксикации (потливость,
- 75. Принципы лечения Хлорбутин или циклофосфан При неэффективности − полихимиотерапия (программы COP, CHOP и др.) Преднизолон при
- 76. В ряде случаев у больных ХЛЛ используются паллиативные методы: лучевая терапия, спленэктомия, лейкацитоферез. Лучевая терапия применяется
- 77. Темпы прогрессирования хронического лимфолейкоза широко варьируют: продолжительность жизни колеблется от 2-3 до 30 лет и более
- 78. «В течение последних двух десятилетий ХЛЛ из неизлечимого заболевания превратился в заболевание, которое в большинстве случаев
- 79. Хронический миелоидный лейкоз – клональное миелопролиферативное заболевание, характеризующееся -- поражением гемопоэза на уровне стволовой кроветворной клетки,
- 80. Впервые ХМЛ был описан немецким патологом R.Virchov (1849)- «селезеночная лейкемия» - первый описанный гемобластоз человека. Частота
- 82. Клеточный состав костного мозга % Клеточный состав костного мозга – 50 – 250 кл в 1
- 83. Этиология ∙ - ионизирующая радиация ( в том числе индуцированная рентгенотерапией), - воздействие бензола. Патогенез У
- 84. Клинические стадии ХМЛ – этапы лейкозного процесса 1 - Хроническая – развернутая (ранняя и поздняя) 2
- 85. Хроническая = развернутая фаза ХМЛ Начальная стадия У большинства пациентов ХМЛ диагностируется в хронической стадии, в
- 86. Основные симптомы: Признаки гиперметаболизма (снижение массы тела, потливость, анорексия); Спленомегалия и ощущения дискомфорта в левом подреберье;
- 87. У больного с ХМЛ увеличение печени, селезенки, геморрагический синдром
- 88. Поздние стадии ХМЛ
- 89. Клинический анализ крови Лейкоцитоз - в среднем более 50х109/л (возможен и низкий уровень лейкоцитов – 15–20х109/л);
- 90. Миелограмма Гиперклеточный костный мозг Гиперплазия нейтрофильного ростка (лейкоэритробластическое соотношение достигает 10-20:1) Количество клеток базофильного и эозинофильного
- 91. Цитохимическим признаком ХМЛ являются снижение активности щелочной фосфатазы нейтрофилов При цитогенетическом исследовании у 95–97% больных выявляется
- 92. На фоне адекватной химиотерапии практически у всех пациентов симптомы исчезают: - значительно уменьшается в размерах или
- 93. фаза акселерации ХМЛ В среднем через 3 года, несмотря на продолжающееся лечение цитостатиками, происходит трансформация заболевания
- 94. Основной лабораторный признак фазы акселерации и бластного криза – прогрессирующее увеличение количества промиелоцитов и бластов в
- 95. Терминальная фаза ХМЛ Обычно терминальная фаза протекает в форме бластного криза В редких случаях ХМЛ впервые
- 96. Критерии диагноза ХМЛ: Лейкоцитоз со сдвигом лейкоцитарной формулы влево Наличие промежуточных форм нейтрофилов Наличие спленомегалии Эозинофильно-базофильная
- 97. Принципы лечения ХМЛ ∙ 1. трансплантация аллогенных стволовых кроветворных клеток ∙ 2. Химиотерапия: миелосан (бусульфан), препаратами
- 98. Трансплантация аллогенных стволовых кроветворных клеток при ХМЛ Аллогенная миелотрансплантация в настоящее время является единственной реальной возможностью
- 99. Аллопрансплантация Костного мозга
- 100. Принципы лечения препаратами гидроксимочевины ∙ Задача врача – снизить и поддерживать уровень лейкоцитов в пределах 5–15х109/л
- 101. ∙ Гливек (иматиниб) специфически ингибирует протеинкиназу, продуцируемую химерным геном BCR-ABL, что приводит к блокаде пролиферации и
- 103. Прогноз при хроническом миелолейкозе Медиана выживаемости больных ХМЛ при стандартной химиотерапии составляет 3,5–4 года. Основные неблагоприятные
- 104. Истинная полицитемия (эритремия) (болезнь Вакеза, polycythemia vera) - доброкачественная опухоль из клетки-предшественницы миелопоэза, сохранившей способность дифференцироваться
- 105. Частота заболевания – 0,6 на 100 тыс населения. Преимущественно болезнь людей старше 50 лет. С одинаковой
- 106. Основные клинические синдромы истинной полицитемии 1) плеторический синдром; 2) генерализованный кожный зуд у 50% больных; эритроцианоз
- 107. Плетора у больных с полицитемией
- 108. Окраска кожи ладоней при эритремии
- 110. Эритремия Вторичная подагра Кожные расчесы
- 111. СТАДИИ ИСТИННОЙ ПОЛИЦИТЕМИИ: 1 – начальная: умеренный эритроцитоз, панмиелоз в костном мозге, небольшое увеличение селезенки. Длительность
- 112. Лабораторные и инструментальные методы Анализ крови: повышение гематокрита >55% у М, > 47% у Ж; Нв
- 113. Лабораторные данные (продолжение) Трепанобиопсия (гиперклеточный костный мозг – 60-100%, при сочетании эритроидной гиперплазии с увеличением мегакариоцитов)
- 114. Современные критерии диагностики истинной полицитемии А1.Увеличение массы циркулирующих эритроцитов более чем на 25% или повышение гемоглобина
- 115. Критерии диагностики истинной полицитемии (окончание) В1. Тромбоцитоз свыше 400 х 109/л В2. Количество лейкоцитов свыше 12
- 118. Лечение истинной полицитемии Начинают стационарно!! 1 Уменьшение объема циркулирующей крови с помощью кровопусканий (250-500мл ч/д) 2-3
- 119. Лечение истинной полицитемии Цитостатики показаны больным пожилого возраста с тяжелыми сердечно-сосудистыми осложнениями, выраженной спленомегалией, лейкоцитозом выше
- 120. Профилактика полицитемии: исключение стрессовых нагрузок, антиаггреганты, малые дозы цитостатиков. Прогноз: продолжительность жизни при адекватном лечении составляет
- 121. Миеломная болезнь (болезнь Рустицкого-Калера)
- 122. Миеломная болезнь – Прогрессирующее, неопластическое заболевание с развитием плазмоклеточных опухолей костного мозга и гиперпродукцией моноклонального иммуноглобулина
- 123. Проявляется обычно у людей после 40 лет. Случаи заболевания в возрасте до 40 лет редки. Частота
- 124. Клиническая картина: ПОРАЖЕНИЕ КОСТЕЙ Разрушение кости при миеломе обусловлено пролиферацией опухолевого клона и активацией остеокластов. При
- 125. Лизис костей приводит к мобилизации кальция из костей и гиперкальциемии с развитием осложнений (тошнота, рвота, сонливость,
- 127. ПОРАЖЕНИЕ ПОЧЕК Миеломная нефропатия. В основе лежит избыточное накопление в канальцах и в строме мозгового, а
- 128. CИНДРОМ ПОВЫШЕННОЙ ВЯЗКОСТИ КРОВИ Развивается при уровне моноклонального белка IgG или IgA выше 50г\л. Клинически проявляется
- 129. НАРУШЕНИЕ ИММУНИТЕТА Высокая частота бактериальных инфекций в связи с гипогаммаглобулинемией, снижением продукции нормальных антител. АНЕМИЯ НОРМОЦИТАРНАЯ
- 130. Диагностика миеломной болезни Классической триадой симптомов миеломной болезни является плазмоцитоз костного мозга (более 10%) сывороточный или
- 131. Миеломные клетки с кристализированным белком Бен-Джонса Миеломные клетки с тальцами Расселя “пламенеющие” (фуксильные) миелоидные клетки
- 132. Мазок нормального красного костного мозга Мазок красного костного мозга при миеломной болезни
- 133. Клинико-лабораторные методы диагностики Биохимические тесты Электрофорез позволяет выявить М-градиент (полосу моноклонового белка в зоне миграции глобулинов
- 134. Лечение миеломной болезни Выбор лечения и его объем зависят от стадии (распространенности) процесса. У 10 %
- 135. Показанием для назначения лечения являются признаки прогрессирования заболевания: отрицательная динамика показателей при повторных исследованиях с интервалом
- 136. Этапы лечения множественной миеломы: I Индукция ремиссии II Период консолидации III Поддерживающее лечение IV Терапия в
- 138. Скачать презентацию

Онкогематологические заболевания (гемобластозы) – злокачественные новообразования, морфологическим субстратом которых являются
Онкогематологические заболевания (гемобластозы) – злокачественные новообразования, морфологическим субстратом которых являются

Максимов А.А. в 1908г впервые высказал представление о наличии единой клетки-предшественницы
Максимов А.А. в 1908г впервые высказал представление о наличии единой клетки-предшественницы


Клеточный состав костного мозга %
Клеточный состав костного мозга – 50 –
Клеточный состав костного мозга %
Клеточный состав костного мозга – 50 –

Распространенность гемобластозов
Суммарная частота гемобластозов: 25–30 случаев на 100 000 населения в
Распространенность гемобластозов
Суммарная частота гемобластозов: 25–30 случаев на 100 000 населения в

Распределение гемобластозов
Все онкогематологические заболевания, за исключением острого лимфобластного лейкоза, лимфобластной лимфомы
Распределение гемобластозов
Все онкогематологические заболевания, за исключением острого лимфобластного лейкоза, лимфобластной лимфомы

Этиологические (предрасполагающие) факторы гемобластозов
В основе - первичная мутации кроветворной клетки-предшественницы под
Этиологические (предрасполагающие) факторы гемобластозов
В основе - первичная мутации кроветворной клетки-предшественницы под

Патогенез онкогематологических заболеваний
Гемобластозы возникают вследствие мутации клетки-предшественницы, приводящей к
Патогенез онкогематологических заболеваний
Гемобластозы возникают вследствие мутации клетки-предшественницы, приводящей к

Патогенез онкогематологических заболеваний (продолжение)
При лабораторных признаках лейкозов в организме больного имеется
Патогенез онкогематологических заболеваний (продолжение)
При лабораторных признаках лейкозов в организме больного имеется

Патогенез онкогематологических заболеваний (окончание)
Для гемобластозов характерна опухолевая прогрессия:
по мере развития
Патогенез онкогематологических заболеваний (окончание)
Для гемобластозов характерна опухолевая прогрессия:
по мере развития

Острые лейкозы – группа клональных заболеваний системы крови, первично поражающих костный
Острые лейкозы – группа клональных заболеваний системы крови, первично поражающих костный

Все острые лейкозы возникают из одной мутировавшей клетки, которая может относиться
Все острые лейкозы возникают из одной мутировавшей клетки, которая может относиться

Острые лейкозы – отдельная группа онкогематологических заболеваний, которые никогда не
Острые лейкозы – отдельная группа онкогематологических заболеваний, которые никогда не

Выделяют два вида заболевания, которые различаются по морфологии, течению, характеру проводимой
Выделяют два вида заболевания, которые различаются по морфологии, течению, характеру проводимой

FAB - классификация острых лейкозов
В 1976 году группой экспертов Франции,
FAB - классификация острых лейкозов
В 1976 году группой экспертов Франции,

Для верификации варианта острого лейкоза и его варианта требуются цитохимические, результаты
Для верификации варианта острого лейкоза и его варианта требуются цитохимические, результаты

Иммунофенотипирование
Внедрение в практику моноклональных антител позволило идентифицировать специфические рецепторы, антигены
Иммунофенотипирование
Внедрение в практику моноклональных антител позволило идентифицировать специфические рецепторы, антигены

Цитогенетическое исследование
У 90% больных ОЛ находят
генетические поломки (транслокации, делеции, инверсии,
Цитогенетическое исследование
У 90% больных ОЛ находят
генетические поломки (транслокации, делеции, инверсии,

Основные клинические синдромы при ОЛ
Синдром лейкемической пролиферации: лимфоаденопатия, гепатоспленомегалия, расширение средостения;
Основные клинические синдромы при ОЛ
Синдром лейкемической пролиферации: лимфоаденопатия, гепатоспленомегалия, расширение средостения;
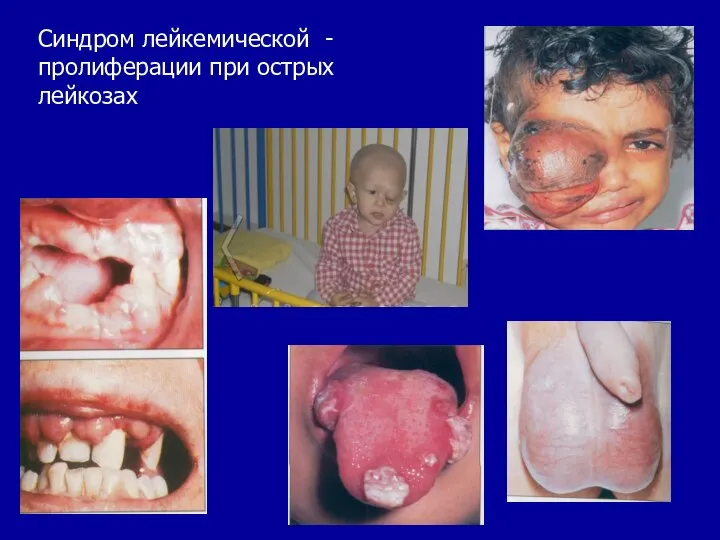
Синдром лейкемической -
пролиферации при острых
лейкозах
Синдром лейкемической -
пролиферации при острых
лейкозах

Проявления геморрагического cиндрома при ОЛ, в том числе
в головной мозг, ЖК-тракт
Инфекционные
Проявления геморрагического cиндрома при ОЛ, в том числе
в головной мозг, ЖК-тракт
Инфекционные

Нейролейкоз
Инфильтрация мозговых оболочек, приведшая к поражению лицевого нерва и одностороннему параличу
Нейролейкоз Инфильтрация мозговых оболочек, приведшая к поражению лицевого нерва и одностороннему параличу

Варианты начала острого лейкоза
- примерно у 50% больных начинается остро
Варианты начала острого лейкоза
- примерно у 50% больных начинается остро

Особенности течения некоторых форм ОМЛ
О недифференцированный лейкоз (М0): частота его уменьшается
Особенности течения некоторых форм ОМЛ
О недифференцированный лейкоз (М0): частота его уменьшается

О. монобластный лейкоз (М5) – 10%, клиника схожа с М4, экстрамедуллярные
О. монобластный лейкоз (М5) – 10%, клиника схожа с М4, экстрамедуллярные

О. Лимфобластный лейкоз – 10-15% (до 25%) взрослых больных ОЛ, (85%
О. Лимфобластный лейкоз – 10-15% (до 25%) взрослых больных ОЛ, (85%

Клинический анализ крови
Количество лейкоцитов периферической крови чаще остается на сублейкемическом
Клинический анализ крови
Количество лейкоцитов периферической крови чаще остается на сублейкемическом

Пример клинического анализа крови больного ОЛ
Гемоглобин – 79г/л;
эритроциты – 2,8×10.12;
Пример клинического анализа крови больного ОЛ
Гемоглобин – 79г/л;
эритроциты – 2,8×10.12;

В типичных случаях между бластами и зрелыми гранулоцитами отсутствуют промежуточные формы
В типичных случаях между бластами и зрелыми гранулоцитами отсутствуют промежуточные формы

Клинический анализ крови-
( морфологическое исследование)
При подсчете лейкоцитарной формулы у 90% больных
Клинический анализ крови-
( морфологическое исследование)
При подсчете лейкоцитарной формулы у 90% больных

Проведение костномозговой пункции обязательно!!!
Иглы для проведения пункции:
Игла Кассирского
Современные иглы
Проведение костномозговой пункции обязательно!!!
Иглы для проведения пункции:
Игла Кассирского
Современные иглы

Техника проведения стернальной пункции
(трепанобиопсии)
Область грудины
Область подвздошной кости
Техника проведения стернальной пункции
(трепанобиопсии)
Область грудины
Область подвздошной кости

Миелограмма- обязательна !
Количество миелокариоцитов обычно повышено, мегакариоциты отсутствуют или их количество
Миелограмма- обязательна !
Количество миелокариоцитов обычно повышено, мегакариоциты отсутствуют или их количество

Для проведения морфологического исследования используются:
1 Клетки периферической крови
2 Мазки-отпечатки лимфоузла (опухоли)
Для проведения морфологического исследования используются:
1 Клетки периферической крови
2 Мазки-отпечатки лимфоузла (опухоли)

Существует правило: необходимо в течение всей жизни больного ОЛ хранить пунктаты
Существует правило: необходимо в течение всей жизни больного ОЛ хранить пунктаты

Стадии острого лейкоза
I. Первая атака (первый острый период) – время
Стадии острого лейкоза
I. Первая атака (первый острый период) – время

Стадии острого лейкоза (окончание)
III. Рецидив – появление клинических и гематологических
Стадии острого лейкоза (окончание)
III. Рецидив – появление клинических и гематологических

Дифференциальный диагноз ОЛ
1 - заболевания, не являющимися гемобластозами:
инфекционный мононуклеоз,
Апластическая
Дифференциальный диагноз ОЛ
1 - заболевания, не являющимися гемобластозами:
инфекционный мононуклеоз,
Апластическая

Прогностические факторы при острых миелоидных лейкозах:
1) Значимые: - возраст более 60
Прогностические факторы при острых миелоидных лейкозах:
1) Значимые: - возраст более 60

Прогностически неблагоприятные факторы при остром лимфобластном лейкозе:
- возраст более 35 лет;
-
Прогностически неблагоприятные факторы при остром лимфобластном лейкозе:
- возраст более 35 лет;
-

Лечение острого лейкоза
1 исторический этап – 19 – начало 20 века
Лечение острого лейкоза
1 исторический этап – 19 – начало 20 века

Принципы химиотерапии острых лейкозов
1. Лечение необходимо начинать сразу же после
Принципы химиотерапии острых лейкозов
1. Лечение необходимо начинать сразу же после

3. Выбор схемы лечения определяется морфологическим, цитохимическим и иммунофенотипическим, цитогенетическим вариантом
3. Выбор схемы лечения определяется морфологическим, цитохимическим и иммунофенотипическим, цитогенетическим вариантом

Основные методы лечения ОЛ
1 Цитостатическая терапия
– уничтожение лейкозного клона;
Основные методы лечения ОЛ
1 Цитостатическая терапия
– уничтожение лейкозного клона;

максимальная эрадикация
лейкозного клона
эрадикация минимальной
резидуальной болезни,
профилактика рецидива – 2-3
максимальная эрадикация
лейкозного клона
эрадикация минимальной
резидуальной болезни,
профилактика рецидива – 2-3

максимальная эрадикация
лейкозного клона
эрадикация минимальной
резидуальной болезни,
профилактика рецидива – 2-3
максимальная эрадикация
лейкозного клона
эрадикация минимальной
резидуальной болезни,
профилактика рецидива – 2-3

Сопроводительная терапия лейкозов:
1 – Дезинтоксикационная терапия (физиологический раствор, полиглюкин, форсированный диурез
Сопроводительная терапия лейкозов:
1 – Дезинтоксикационная терапия (физиологический раствор, полиглюкин, форсированный диурез

Осложнения острых лейкозов
1. Инфекционные осложнения (у 70% больных определяют летальные исходы).
Для
Осложнения острых лейкозов
1. Инфекционные осложнения (у 70% больных определяют летальные исходы).
Для

2 Геморрагический синдром
- выраженная тромбоцитопения – у 1\2 больных
- летальность от
2 Геморрагический синдром
- выраженная тромбоцитопения – у 1\2 больных
- летальность от

3 Анемический синдром
- анемия встречается у всех больных ОЛ,
- основной
3 Анемический синдром
- анемия встречается у всех больных ОЛ,
- основной

4 Энтеропатия
Состояние, характеризующееся поражением слизистой ЖКТ, возникающее на фоне миелотоксического агранулоцитоза
4 Энтеропатия
Состояние, характеризующееся поражением слизистой ЖКТ, возникающее на фоне миелотоксического агранулоцитоза

6 Нейролейкоз
- частота при ОЛЛ – 30-50%; при ОНЛЛ- 5%.
Клинические варианты:
6 Нейролейкоз
- частота при ОЛЛ – 30-50%; при ОНЛЛ- 5%.
Клинические варианты:

Современная химиотерапия позволяет получить полные ремиссии у 50–95% взрослых больных
В
Современная химиотерапия позволяет получить полные ремиссии у 50–95% взрослых больных
В

Лекция
для 5 курса 4 факультета ФПВ на тему:
« Хронические лейкозы
Лекция
для 5 курса 4 факультета ФПВ на тему:
« Хронические лейкозы

Хронические лейкозы – гетерогенная группа опухолевых заболеваний крови, морфологическим субстратом которых
Хронические лейкозы – гетерогенная группа опухолевых заболеваний крови, морфологическим субстратом которых

Хронический лимфолейкоз (хронический лимфоцитарный лейкоз - клональное лимфопролиферативное заболевание, первично локализующееся
Хронический лимфолейкоз (хронический лимфоцитарный лейкоз - клональное лимфопролиферативное заболевание, первично локализующееся


Гемопоэз
Гемопоэз

Эпидемиология
Составляет более 30% всех лейкозов в Северной Америке и Европе
Ежегодная
Эпидемиология
Составляет более 30% всех лейкозов в Северной Америке и Европе
Ежегодная

Этиология ХЛЛ
Для ХЛЛ не доказана этиологическая роль ионизирующего облучения.
Этиология ХЛЛ
Для ХЛЛ не доказана этиологическая роль ионизирующего облучения.
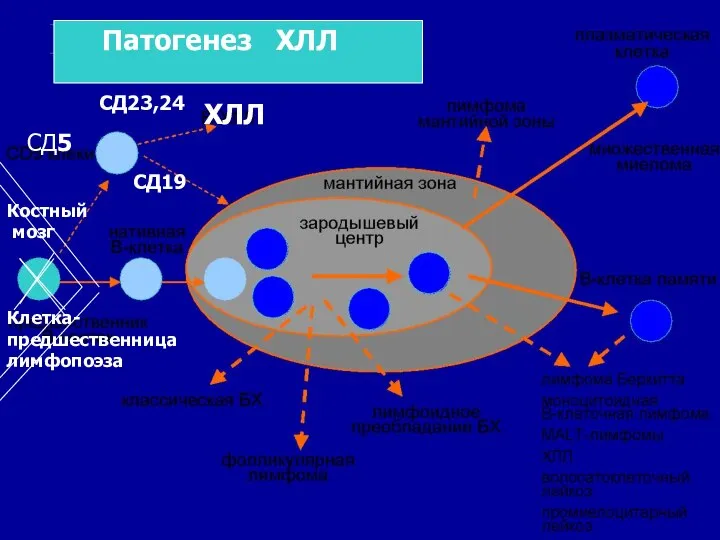
ХЛЛ
СД5
Клетка-предшественница лимфопоэза
Костный
мозг
СД19
СД23,24
Патогенез ХЛЛ
ХЛЛ
СД5
Клетка-предшественница лимфопоэза
Костный
мозг
СД19
СД23,24
Патогенез ХЛЛ

Патогенетические особенности ХЛЛ:
- пролиферация клона трансформированных «зрелых» лимфоцитов;
в иммунофенотипе -
Патогенетические особенности ХЛЛ:
- пролиферация клона трансформированных «зрелых» лимфоцитов;
в иммунофенотипе -

Клиническая картина
В 70% случаев заболевание начинается постепенно и характеризуется лишь абсолютным
Клиническая картина
В 70% случаев заболевание начинается постепенно и характеризуется лишь абсолютным

Синдром лимфатической
пролиферации при ХЛЛ
Синдром лимфатической
пролиферации при ХЛЛ

2 Симптомы опухолевой интоксикации: истощение, боли в костях, повышение температуры тела.
3
2 Симптомы опухолевой интоксикации: истощение, боли в костях, повышение температуры тела.
3

Лабораторные данные
Лейкоцитоз с абсолютным (более 5 × 109/л)
и относительным лимфоцитозом;
Морфологически
Лабораторные данные
Лейкоцитоз с абсолютным (более 5 × 109/л)
и относительным лимфоцитозом;
Морфологически

Нормальные показатели клинического анализа крови
Нормальные показатели клинического анализа крови

Клинический анализ крови при ХЛЛ
Клинический анализ крови при ХЛЛ

Миелограмма
1) При ХЛЛ;
2) нормальная
Миелограмма
1) При ХЛЛ;
2) нормальная


Критерии диагноза
Абсолютный лимфоцитоз более 5 (10) 109/л
Нормальная морфология лимфоцитов при световой
Критерии диагноза
Абсолютный лимфоцитоз более 5 (10) 109/л
Нормальная морфология лимфоцитов при световой
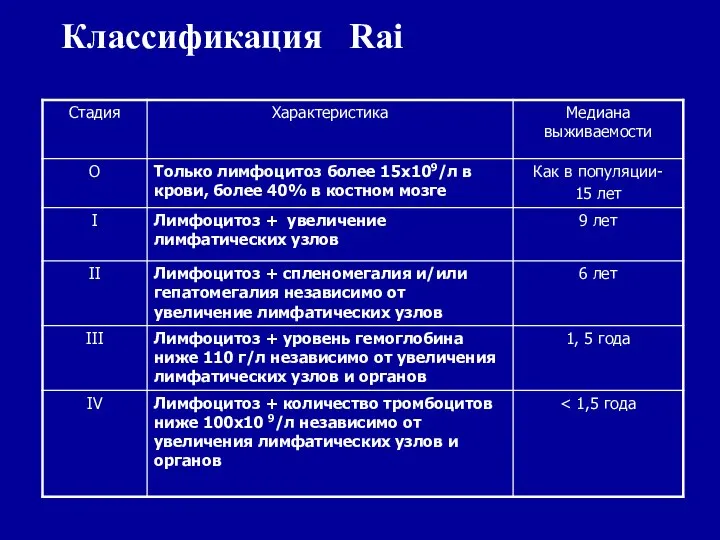
Классификация Rai
Классификация Rai

Критерии для начала лечения
(Рано начатое лечение не увеличивает продолжительность жизни больных
Критерии для начала лечения
(Рано начатое лечение не увеличивает продолжительность жизни больных

Принципы лечения
Хлорбутин или циклофосфан
При неэффективности − полихимиотерапия (программы COP, CHOP
Принципы лечения
Хлорбутин или циклофосфан
При неэффективности − полихимиотерапия (программы COP, CHOP

В ряде случаев у больных ХЛЛ используются паллиативные методы: лучевая терапия,
В ряде случаев у больных ХЛЛ используются паллиативные методы: лучевая терапия,

Темпы прогрессирования хронического лимфолейкоза широко варьируют: продолжительность жизни колеблется от 2-3
Темпы прогрессирования хронического лимфолейкоза широко варьируют: продолжительность жизни колеблется от 2-3

«В течение последних двух десятилетий ХЛЛ из неизлечимого заболевания превратился
«В течение последних двух десятилетий ХЛЛ из неизлечимого заболевания превратился

Хронический миелоидный лейкоз
– клональное миелопролиферативное заболевание, характеризующееся
-- поражением гемопоэза на
Хронический миелоидный лейкоз – клональное миелопролиферативное заболевание, характеризующееся -- поражением гемопоэза на

Впервые ХМЛ был описан немецким патологом R.Virchov (1849)- «селезеночная лейкемия»
Впервые ХМЛ был описан немецким патологом R.Virchov (1849)- «селезеночная лейкемия»


Клеточный состав костного мозга %
Клеточный состав костного мозга – 50 –
Клеточный состав костного мозга %
Клеточный состав костного мозга – 50 –

Этиология
∙ - ионизирующая радиация ( в том числе индуцированная рентгенотерапией),
-
Этиология
∙ - ионизирующая радиация ( в том числе индуцированная рентгенотерапией),
-

Клинические стадии ХМЛ – этапы лейкозного процесса
1 - Хроническая – развернутая
Клинические стадии ХМЛ – этапы лейкозного процесса
1 - Хроническая – развернутая

Хроническая = развернутая фаза ХМЛ
Начальная стадия
У большинства пациентов ХМЛ диагностируется
Хроническая = развернутая фаза ХМЛ
Начальная стадия
У большинства пациентов ХМЛ диагностируется

Основные симптомы:
Признаки гиперметаболизма (снижение массы тела, потливость, анорексия);
Спленомегалия и ощущения дискомфорта
Основные симптомы:
Признаки гиперметаболизма (снижение массы тела, потливость, анорексия);
Спленомегалия и ощущения дискомфорта

У больного с ХМЛ
увеличение печени,
селезенки, геморрагический
синдром
У больного с ХМЛ
увеличение печени,
селезенки, геморрагический
синдром

Поздние стадии ХМЛ
Поздние стадии ХМЛ

Клинический анализ крови
Лейкоцитоз - в среднем более 50х109/л (возможен и
Клинический анализ крови
Лейкоцитоз - в среднем более 50х109/л (возможен и

Миелограмма
Гиперклеточный костный мозг
Гиперплазия нейтрофильного ростка (лейкоэритробластическое соотношение достигает
10-20:1)
Миелограмма
Гиперклеточный костный мозг
Гиперплазия нейтрофильного ростка (лейкоэритробластическое соотношение достигает
10-20:1)

Цитохимическим признаком ХМЛ являются снижение активности щелочной фосфатазы нейтрофилов
При цитогенетическом исследовании
Цитохимическим признаком ХМЛ являются снижение активности щелочной фосфатазы нейтрофилов
При цитогенетическом исследовании

На фоне адекватной химиотерапии практически у всех пациентов симптомы исчезают:
-
На фоне адекватной химиотерапии практически у всех пациентов симптомы исчезают:
-

фаза акселерации ХМЛ
В среднем через 3 года, несмотря на продолжающееся
фаза акселерации ХМЛ
В среднем через 3 года, несмотря на продолжающееся

Основной лабораторный признак фазы акселерации и бластного криза – прогрессирующее увеличение
Основной лабораторный признак фазы акселерации и бластного криза – прогрессирующее увеличение

Терминальная фаза ХМЛ
Обычно терминальная фаза протекает в форме бластного криза
В
Терминальная фаза ХМЛ
Обычно терминальная фаза протекает в форме бластного криза
В

Критерии диагноза ХМЛ:
Лейкоцитоз со сдвигом лейкоцитарной формулы влево
Наличие промежуточных форм
Критерии диагноза ХМЛ:
Лейкоцитоз со сдвигом лейкоцитарной формулы влево
Наличие промежуточных форм

Принципы лечения ХМЛ
∙ 1. трансплантация аллогенных стволовых кроветворных клеток
∙ 2. Химиотерапия: миелосан (бусульфан),
Принципы лечения ХМЛ
∙ 1. трансплантация аллогенных стволовых кроветворных клеток
∙ 2. Химиотерапия: миелосан (бусульфан),

Трансплантация аллогенных стволовых кроветворных клеток при ХМЛ
Аллогенная миелотрансплантация в настоящее время
Трансплантация аллогенных стволовых кроветворных клеток при ХМЛ
Аллогенная миелотрансплантация в настоящее время
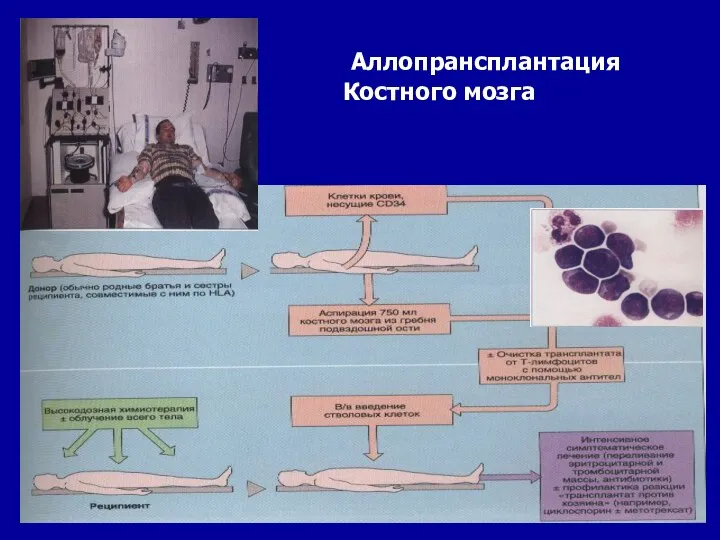
Аллопрансплантация
Костного мозга
Аллопрансплантация
Костного мозга

Принципы лечения препаратами гидроксимочевины
∙ Задача врача – снизить и поддерживать уровень
Принципы лечения препаратами гидроксимочевины
∙ Задача врача – снизить и поддерживать уровень

∙ Гливек (иматиниб)
специфически ингибирует протеинкиназу, продуцируемую химерным геном BCR-ABL, что
∙ Гливек (иматиниб) специфически ингибирует протеинкиназу, продуцируемую химерным геном BCR-ABL, что


Прогноз при хроническом миелолейкозе
Медиана выживаемости больных ХМЛ при стандартной химиотерапии составляет
Прогноз при хроническом миелолейкозе
Медиана выживаемости больных ХМЛ при стандартной химиотерапии составляет

Истинная полицитемия (эритремия)
(болезнь Вакеза, polycythemia vera)
- доброкачественная опухоль из клетки-предшественницы миелопоэза,
Истинная полицитемия (эритремия)
(болезнь Вакеза, polycythemia vera)
- доброкачественная опухоль из клетки-предшественницы миелопоэза,

Частота заболевания – 0,6 на 100 тыс населения.
Преимущественно болезнь людей старше
Частота заболевания – 0,6 на 100 тыс населения.
Преимущественно болезнь людей старше

Основные клинические синдромы истинной полицитемии
1) плеторический синдром;
2) генерализованный кожный
Основные клинические синдромы истинной полицитемии
1) плеторический синдром;
2) генерализованный кожный
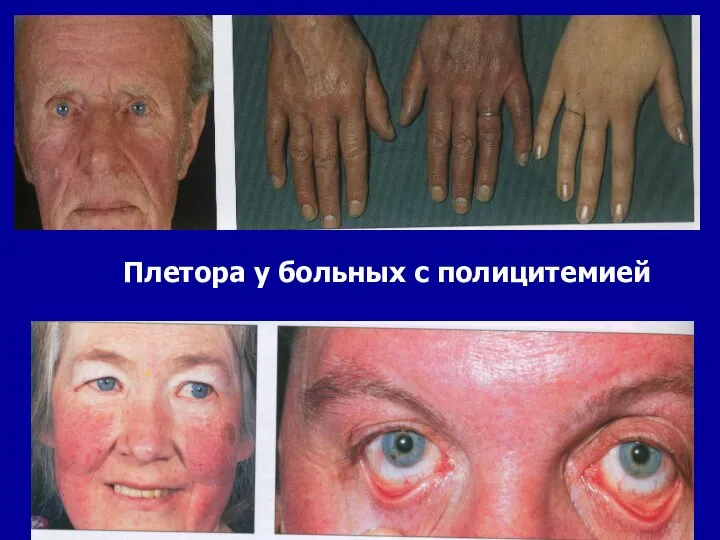
Плетора у больных с полицитемией

Окраска кожи ладоней при эритремии
Окраска кожи ладоней при эритремии


Эритремия
Вторичная
подагра
Кожные расчесы
Эритремия
Вторичная
подагра
Кожные расчесы

СТАДИИ ИСТИННОЙ ПОЛИЦИТЕМИИ:
1 – начальная: умеренный эритроцитоз, панмиелоз в костном мозге,
СТАДИИ ИСТИННОЙ ПОЛИЦИТЕМИИ:
1 – начальная: умеренный эритроцитоз, панмиелоз в костном мозге,

Лабораторные и инструментальные методы
Анализ крови: повышение гематокрита >55% у М, >
Лабораторные и инструментальные методы
Анализ крови: повышение гематокрита >55% у М, >

Лабораторные данные (продолжение)
Трепанобиопсия (гиперклеточный костный мозг – 60-100%, при сочетании эритроидной
Лабораторные данные (продолжение)
Трепанобиопсия (гиперклеточный костный мозг – 60-100%, при сочетании эритроидной

Современные критерии диагностики истинной полицитемии
А1.Увеличение массы циркулирующих эритроцитов более чем на
Современные критерии диагностики истинной полицитемии
А1.Увеличение массы циркулирующих эритроцитов более чем на

Критерии диагностики истинной полицитемии (окончание)
В1. Тромбоцитоз свыше 400 х 109/л
В2. Количество
Критерии диагностики истинной полицитемии (окончание)
В1. Тромбоцитоз свыше 400 х 109/л
В2. Количество



Лечение истинной полицитемии
Начинают стационарно!!
1 Уменьшение объема циркулирующей крови с помощью кровопусканий
Лечение истинной полицитемии
Начинают стационарно!!
1 Уменьшение объема циркулирующей крови с помощью кровопусканий

Лечение истинной полицитемии
Цитостатики показаны больным пожилого возраста с тяжелыми сердечно-сосудистыми осложнениями,
Лечение истинной полицитемии
Цитостатики показаны больным пожилого возраста с тяжелыми сердечно-сосудистыми осложнениями,

Профилактика полицитемии: исключение стрессовых нагрузок, антиаггреганты, малые дозы цитостатиков.
Прогноз:
продолжительность
Профилактика полицитемии: исключение стрессовых нагрузок, антиаггреганты, малые дозы цитостатиков.
Прогноз:
продолжительность

Миеломная болезнь
(болезнь Рустицкого-Калера)
Миеломная болезнь
(болезнь Рустицкого-Калера)

Миеломная болезнь –
Прогрессирующее, неопластическое заболевание с развитием плазмоклеточных опухолей костного
Миеломная болезнь –
Прогрессирующее, неопластическое заболевание с развитием плазмоклеточных опухолей костного

Проявляется обычно у людей после 40 лет.
Случаи заболевания в возрасте
Проявляется обычно у людей после 40 лет.
Случаи заболевания в возрасте

Клиническая картина: ПОРАЖЕНИЕ КОСТЕЙ
Разрушение кости при миеломе обусловлено пролиферацией опухолевого клона
Клиническая картина: ПОРАЖЕНИЕ КОСТЕЙ
Разрушение кости при миеломе обусловлено пролиферацией опухолевого клона

Лизис костей приводит к мобилизации кальция из костей и гиперкальциемии с
Лизис костей приводит к мобилизации кальция из костей и гиперкальциемии с


ПОРАЖЕНИЕ ПОЧЕК
Миеломная нефропатия. В основе лежит избыточное накопление в канальцах
ПОРАЖЕНИЕ ПОЧЕК
Миеломная нефропатия. В основе лежит избыточное накопление в канальцах
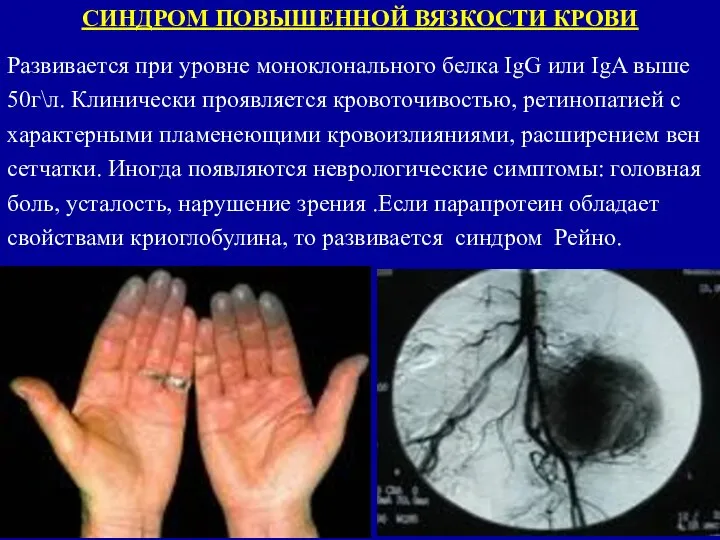
CИНДРОМ ПОВЫШЕННОЙ ВЯЗКОСТИ КРОВИ
Развивается при уровне моноклонального белка IgG или IgA
CИНДРОМ ПОВЫШЕННОЙ ВЯЗКОСТИ КРОВИ
Развивается при уровне моноклонального белка IgG или IgA

НАРУШЕНИЕ ИММУНИТЕТА
Высокая частота бактериальных инфекций в связи с гипогаммаглобулинемией, снижением продукции
НАРУШЕНИЕ ИММУНИТЕТА
Высокая частота бактериальных инфекций в связи с гипогаммаглобулинемией, снижением продукции

Диагностика миеломной болезни
Классической триадой симптомов миеломной болезни является
плазмоцитоз костного мозга
Диагностика миеломной болезни
Классической триадой симптомов миеломной болезни является
плазмоцитоз костного мозга

Миеломные клетки с кристализированным белком Бен-Джонса
Миеломные клетки с тальцами Расселя
“пламенеющие” (фуксильные)
Миеломные клетки с кристализированным белком Бен-Джонса
Миеломные клетки с тальцами Расселя
“пламенеющие” (фуксильные)

Мазок нормального красного костного мозга
Мазок красного костного мозга при миеломной болезни
Мазок нормального красного костного мозга
Мазок красного костного мозга при миеломной болезни

Клинико-лабораторные методы диагностики
Биохимические тесты
Электрофорез позволяет выявить М-градиент (полосу моноклонового белка в
Клинико-лабораторные методы диагностики
Биохимические тесты
Электрофорез позволяет выявить М-градиент (полосу моноклонового белка в

Лечение миеломной болезни
Выбор лечения и его объем зависят от стадии (распространенности)
Лечение миеломной болезни
Выбор лечения и его объем зависят от стадии (распространенности)

Показанием для назначения лечения являются признаки прогрессирования заболевания:
отрицательная динамика показателей при
Показанием для назначения лечения являются признаки прогрессирования заболевания:
отрицательная динамика показателей при

Этапы лечения множественной миеломы:
I Индукция ремиссии
II Период консолидации
III Поддерживающее лечение
IV Терапия
Этапы лечения множественной миеломы:
I Индукция ремиссии
II Период консолидации
III Поддерживающее лечение
IV Терапия
 Источники питания недоношенных детей. Режимы кормления
Источники питания недоношенных детей. Режимы кормления Болеутоляющие (анальгезирующие) средства
Болеутоляющие (анальгезирующие) средства Всемирный день памяти людей, умерших от СПИДа
Всемирный день памяти людей, умерших от СПИДа Арт-терапия в работе с психосоматическими расстройствами
Арт-терапия в работе с психосоматическими расстройствами Сумен емдеу
Сумен емдеу Я, мы, они
Я, мы, они Кесарево сечение в современном акушерстве и родовой травматизм
Кесарево сечение в современном акушерстве и родовой травматизм Паразитарные хирургические заболевания
Паразитарные хирургические заболевания Стратегия и приоритеты развития здравоохранения
Стратегия и приоритеты развития здравоохранения Плазменное омоложение лица
Плазменное омоложение лица Артропластика плечевого, локтевого, тазобедренного, коленного суставов, артродез, эндопротезирование суставов
Артропластика плечевого, локтевого, тазобедренного, коленного суставов, артродез, эндопротезирование суставов Интоксикации пестицидами
Интоксикации пестицидами Психология ранней юности
Психология ранней юности Возбудители атипичных пневмоний
Возбудители атипичных пневмоний Угрозы сексуального поведения
Угрозы сексуального поведения Резекция желудка по Бильрот
Резекция желудка по Бильрот Сальмонеллёзы. Клиническая картина
Сальмонеллёзы. Клиническая картина Личность. Структура личности
Личность. Структура личности Хирургическая анатомия лицевого отдела головы, операции на лицевом отделе головы
Хирургическая анатомия лицевого отдела головы, операции на лицевом отделе головы Системная склеродермия
Системная склеродермия Лечение больных немелкоклеточным раком легкого
Лечение больных немелкоклеточным раком легкого Лечение хронической обструктивной болезни легких (ХОБЛ)
Лечение хронической обструктивной болезни легких (ХОБЛ) Головная боль
Головная боль Острый и хронический гломерулонефрит
Острый и хронический гломерулонефрит Снотворные средства
Снотворные средства Патология женских и мужских половых желез: основные виды, характеристика, проявления, принципы терапии
Патология женских и мужских половых желез: основные виды, характеристика, проявления, принципы терапии Введение в спланхнологию. Закономерности развития и строения пищеварительной системы. Особенности у детей
Введение в спланхнологию. Закономерности развития и строения пищеварительной системы. Особенности у детей Психология развития
Психология развития