- Сахарный диабет

Содержание
- 2. Значимость проблемы Среди эндокринной патологии СД занимает первое место (> 50% всех эндокринных заболеваний). Сегодня диабетом
- 3. Значимость проблемы (продолжение) Каждые 10 секунд в мире: - становится на 2 больных больше - ≈
- 4. Классификация СД по тяжести течения I. Лёгкое течение: - невысокий уровень гликемии (не > 8 ммоль/л
- 5. Классификация СД по степени компенсации 1. Компенсированная форма: - нормальные показатели глюкозы в крови и -
- 6. Классификация СД (этиопатогенетическая) I. СД 1-го типа. II. СД 2-го типа. III. Другие специфические формы СД.
- 7. СД 1-го типа Заболевание, которое развивается вследствие прогрессирующего разрушения β-клеток островков Лангерганса поджелудочной железы, сопровождающееся дефицитом
- 8. Этиология СД 1 Основной этиологический фактор – изменение АГ состава β-клеток островкового аппарата поджелудочной железы, возникающее
- 9. Этиология СД 1 (продолжение) Генетическая предрасположенность обусловлена несколькими генами, в том числе дефектом генов системы HLA
- 10. Этиология СД 1 (продолжение) Аллели генов HLA могут обусловливать: • предрасположенность: - аллели HLA-DR3, -DR4 либо
- 11. Этиология СД 1 (продолжение) Наличие наследственной предрасположенности свидетельствует о большей вероятности развития СД1, чем у тех,
- 12. Этиология СД 1 (продолжение) Особое значение имеют β-цитотропные вирусы: • краснухи (имеет тропизм к островкам pancreas,
- 13. Патогенез СД 1 Главное звено патогенеза - разрушение β-клеток. Имеет аутоиммунную природу. Участвуют Гуморальное звено Клеточное
- 14. Патогенез СД 1 (продолжение) Механизм гибели β-клеток Генетическая предрасположенность к диабету + вирусная инфекция: → изменение
- 15. Патогенез СД 1 (продолжение)
- 16. Патогенез СД 1 (продолжение) Примечание: IA - АТ к инсулину, GAD - АТ к глутаматдекабоксилазе
- 17. Характеристика СД 1 типа 1. Начало болезни – в молодом, или – чаще – в детском
- 18. Роль инсулина в организме Роль инсулина в организме исключительна. Основное действие инсулина: ● на углеводный обмен:
- 19. Лечение СД 1 типа I. Консервативная терапия. 1. Пожизненная заместительная инсулинотерапия - подбор базового уровня продлённых
- 20. Этиология СД 2 типа СД2 – самая частая форма СД - ≈ 90% всех случаев. СД2
- 21. Этиология СД2 типа (продолжение) Определенные сочетания генов обусловливают предрасположенность к болезни, а ее развитие и клиническое
- 22. Патогенез СД 2 типа У больных СД 2 типа нет ↓ числа β-клеток. Патология не связана
- 23. Патогенез СД 2 (продолжение) 2. Пострецепторная ИР - нарушение пострецепторных механизмов, опосредующих эффекты инсулина. Пострецепторные дефекты
- 24. Патогенез СД 2 типа
- 25. Патогенез СД 2 (продолжение) II. Наследственное нарушение ранней секреции инсулина β-клетками - ↓ первая фаза секреции
- 26. Ранняя фаза выделения инсулина
- 27. Патогенез СД 2 (продолжение) Механизмы нарушения ранней секреции 1. Генетический дефект механизма восприятия β-клетками ↑ уровня
- 28. Патогенез СД 2 (продолжение) Механизмы нарушения ранней секреции 2. Наследственное ↓ продукции ГПП-1 (глюкагоноподобный пептид). ГПП-1
- 29. Характеристика СД 2 типа 1. Начало заболевания - в зрелом или пожилом возрасте (бывает и у
- 30. Лечение СД 2-го типа 1. Немедикаментозная терапия Основа – постулат: «Диабет - не болезнь, а образ
- 31. Лечение СД 2-го типа (продолжение) 2. Медикаментозная терапия • Препараты, снижающие всасываемость глюкозы в кишечнике (угнетают
- 32. Хирургическое лечение СД 2 типа 1. Метаболическая хирургия: - желудочное (гастрошунтирование) и - билиопанкреатическое шунтирование (пища
- 33. Отличия СД 1 и СД2
- 34. Гестационный СД Возникает при беременности. Может полностью исчезнуть после родов. ГСД - нарушение толерантности к глюкозе,
- 35. Гестационный СД Влияет на: • плод: - внутриутробная гибель плода (из-за кетоза или поражения сосудов плаценты
- 36. Другие специфические формы СД 1) СД при: - органических поражениях поджелудочной железы (хронический панкреатит, панкреатэктомия (в
- 37. Другие специфические формы СД (продолжение) 2) СД, вызванный гиперпродукцией сахароповышающих гормонов (глюкагон, КА, СТГ, АКТГ, глюкокортикоиды)
- 38. Другие специфические формы СД (продолжение) 3) СД, вызванный: • химическими веществами (многие из них нарушают секрецию
- 39. Другие специфические формы СД (продолжение) 4) Моногенные формы инсулинонезависимого СД - обусловлены мутациями генов, контролирующих секрецию
- 40. Лабораторные и клинические проявления СД I. Нарушения углеводного обмена 1. Гипергликемия - ↑ уровня глюкозы в
- 41. Лабораторные и клинические проявления СД (продолжение) 2. Глюкозурия – появление глюкозы в моче. Механизм. У здорового
- 42. Лабораторные и клинические проявления СД (продолжение) 3. Нарушение толерантности к глюкозе – ↓ устойчивости больных к
- 43. Глюкозотолерантный тест
- 44. Другие критерии диагностики СД 4. ↑ содержания в крови гликозилированного (гликированного) Hb (HbА1: a, b, c).
- 45. Диагностика гестационного СД Заподозрить развитие у женщины СД беременных можно по анализу крови, в котором уровень
- 46. Нарушения липидного обмена Лучше изучены для СД 1 типа. Абсолютный дефицит инсулина нарушает обмен липидов на
- 47. Нарушения липидного обмена (продолжение) Расщепление ТГ формирует большой объем СЖК → выбрасываются в кровоток → СЖК
- 48. Нарушения липидного обмена (продолжение) 2. Большая часть окисляется до ацетил-КоА: из-за нарушения утилизации АцКоА в цикле
- 49. Нарушения белкового обмена Инсулин – мощный анаболический гормон (стимулирует синтез нуклеиновых кислот, ↑ синтез белка -
- 50. Диабетическая стопа Поражение стоп больного в виде гнойно-некротических процессов, язв и костно-суставных поражений, возникающее на фоне
- 51. Водно-электролитный обмен Экскреция кетоновых тел почками уменьшает содержание в организме связанных оснований, что ведет к дополнительной
- 52. Классификация осложнений I. Острые – кома: • собственно диабетическая: - кетоацидотическая, - гиперосмолярная, - лактацидотическая, •
- 53. Кетоацидотическая кома Обусловлена токсическим влиянием кетоновых тел на клетки ЦНС (блокируют выработку энергии в нейронах мозга,
- 54. Гиперосмолярная кома Встречается главным образом у пожилых больных с СД 2 типа. Механизм Обезвоживание клеток ЦНС
- 55. Лактацидотическая кома Чаще возникает у больных старше 50 лет на фоне сердечно-сосудистой, печеночной и почечной недостаточности,
- 56. Гипогликемическая кома Этиология • передозировка инсулина или пероральных сахароснижающих препаратов, • нарушение диеты (несвоевременность или прием
- 57. Микроангиопатии М. – это комплекс специфических (характерны только для СД) патологических изменений в сосудах микроциркуляции (артериолы,
- 58. Микроангиопатии (продолжение) Механизмы микроангиопатий Главный патогенетический фактор - хроническая гипергликемия - индуцирует ряд патологических механизмов (каскад
- 59. Механизмы микроангиопатий (продолжение) • повышенное аутоокисление глюкозы → активация процессов ПОЛ → накопление свободных радикалов (→
- 60. Диабетические макроангиопатии МА - поражения артерий крупного и среднего калибра, с образованием на их эндотелии атеросклеротических
- 61. Макроангиопатии при СД (продолжение) Диабетические МА (в соответствии с локализацией и клиническими проявлениями) подразделяются на: поражения
- 62. Макроангиопатии при СД (продолжение) Дополнительно: • ↓ ЛПВП + ↑ ЛПНП (нарушения липидного обмена + АГ
- 63. Профилактика осложнений Контроль АД. Преимущество отдаётся: - метаболически нейтральным (ИАПФ, сартаны) и - метаболически позитивным (моксонидин)
- 65. Скачать презентацию

Значимость проблемы
Среди эндокринной патологии СД занимает первое место (> 50% всех
Значимость проблемы
Среди эндокринной патологии СД занимает первое место (> 50% всех

Значимость проблемы (продолжение)
Каждые 10 секунд в мире:
- становится на 2 больных
Значимость проблемы (продолжение)
Каждые 10 секунд в мире:
- становится на 2 больных

Классификация СД по тяжести течения
I. Лёгкое течение:
- невысокий уровень
Классификация СД по тяжести течения
I. Лёгкое течение:
- невысокий уровень
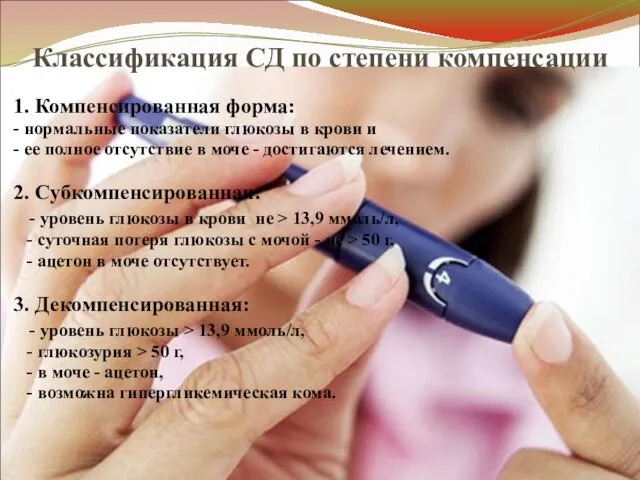
Классификация СД по степени компенсации
1. Компенсированная форма:
- нормальные показатели глюкозы
Классификация СД по степени компенсации
1. Компенсированная форма:
- нормальные показатели глюкозы

Классификация СД
(этиопатогенетическая)
I. СД 1-го типа.
II. СД 2-го типа.
III. Другие специфические
Классификация СД
(этиопатогенетическая)
I. СД 1-го типа.
II. СД 2-го типа.
III. Другие специфические

СД 1-го типа
Заболевание, которое развивается вследствие прогрессирующего разрушения
β-клеток островков Лангерганса
СД 1-го типа
Заболевание, которое развивается вследствие прогрессирующего разрушения
β-клеток островков Лангерганса

Этиология СД 1
Основной этиологический фактор
– изменение АГ состава β-клеток
островкового
Этиология СД 1
Основной этиологический фактор
– изменение АГ состава β-клеток
островкового

Этиология СД 1 (продолжение)
Генетическая предрасположенность обусловлена несколькими генами, в том числе
Этиология СД 1 (продолжение)
Генетическая предрасположенность обусловлена несколькими генами, в том числе

Этиология СД 1 (продолжение)
Аллели генов HLA могут обусловливать:
• предрасположенность:
- аллели HLA-DR3,
Этиология СД 1 (продолжение)
Аллели генов HLA могут обусловливать:
• предрасположенность:
- аллели HLA-DR3,

Этиология СД 1 (продолжение)
Наличие наследственной предрасположенности свидетельствует о
большей вероятности развития
Этиология СД 1 (продолжение)
Наличие наследственной предрасположенности свидетельствует о
большей вероятности развития

Этиология СД 1 (продолжение)
Особое значение имеют β-цитотропные вирусы:
• краснухи (имеет
Этиология СД 1 (продолжение)
Особое значение имеют β-цитотропные вирусы:
• краснухи (имеет
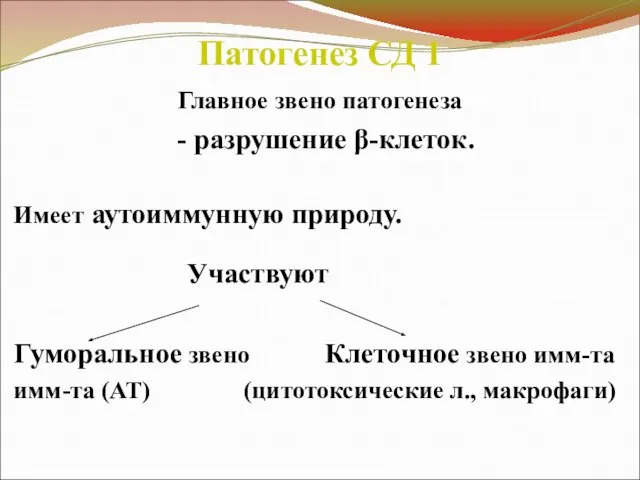
Патогенез СД 1
Главное звено патогенеза
- разрушение β-клеток.
Имеет аутоиммунную природу.
Участвуют
Гуморальное
Патогенез СД 1
Главное звено патогенеза
- разрушение β-клеток.
Имеет аутоиммунную природу.
Участвуют
Гуморальное

Патогенез СД 1 (продолжение)
Механизм гибели β-клеток
Генетическая предрасположенность к диабету +
Патогенез СД 1 (продолжение)
Механизм гибели β-клеток
Генетическая предрасположенность к диабету +
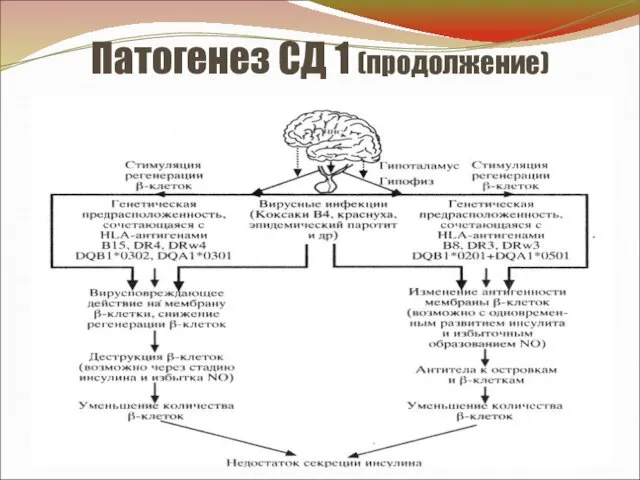
Патогенез СД 1 (продолжение)
Патогенез СД 1 (продолжение)
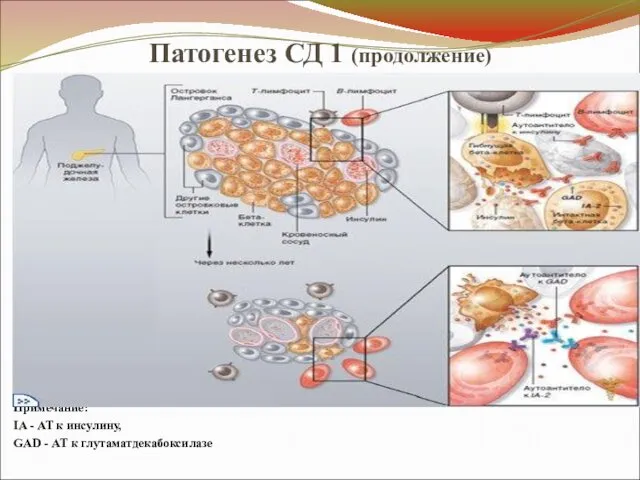
Патогенез СД 1 (продолжение)
Примечание:
IA - АТ к инсулину,
GAD - АТ к
Патогенез СД 1 (продолжение)
Примечание:
IA - АТ к инсулину,
GAD - АТ к

Характеристика СД 1 типа
1. Начало болезни – в молодом, или –
Характеристика СД 1 типа
1. Начало болезни – в молодом, или –

Роль инсулина в организме
Роль инсулина в организме исключительна.
Основное действие инсулина:
●
Роль инсулина в организме
Роль инсулина в организме исключительна.
Основное действие инсулина:
●

Лечение СД 1 типа
I. Консервативная терапия.
1. Пожизненная заместительная инсулинотерапия - подбор
Лечение СД 1 типа
I. Консервативная терапия.
1. Пожизненная заместительная инсулинотерапия - подбор

Этиология СД 2 типа
СД2 – самая частая форма СД - ≈
Этиология СД 2 типа
СД2 – самая частая форма СД - ≈

Этиология СД2 типа (продолжение)
Определенные сочетания генов обусловливают предрасположенность к болезни, а ее развитие
Этиология СД2 типа (продолжение)
Определенные сочетания генов обусловливают предрасположенность к болезни, а ее развитие

Патогенез СД 2 типа
У больных СД 2 типа нет ↓ числа
Патогенез СД 2 типа
У больных СД 2 типа нет ↓ числа

Патогенез СД 2 (продолжение)
2. Пострецепторная ИР - нарушение пострецепторных механизмов, опосредующих
Патогенез СД 2 (продолжение)
2. Пострецепторная ИР - нарушение пострецепторных механизмов, опосредующих
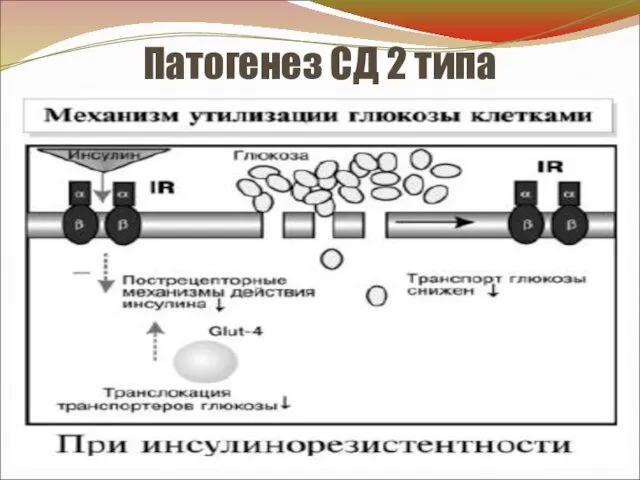
Патогенез СД 2 типа
Патогенез СД 2 типа

Патогенез СД 2 (продолжение)
II. Наследственное нарушение ранней секреции инсулина β-клетками
-
Патогенез СД 2 (продолжение)
II. Наследственное нарушение ранней секреции инсулина β-клетками
-
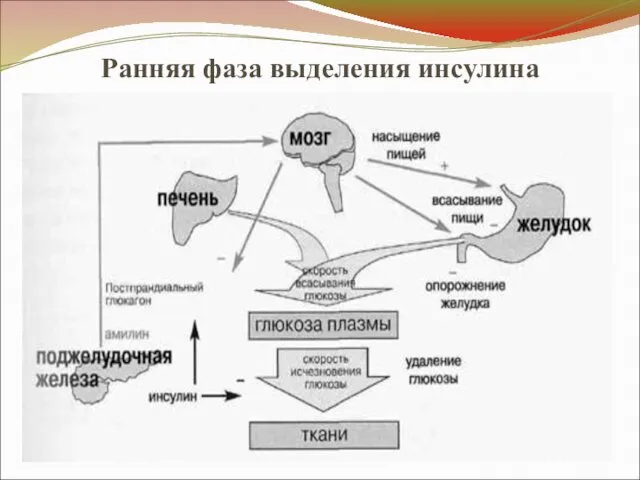
Ранняя фаза выделения инсулина
Ранняя фаза выделения инсулина

Патогенез СД 2 (продолжение)
Механизмы нарушения ранней секреции
1. Генетический дефект механизма восприятия
Патогенез СД 2 (продолжение)
Механизмы нарушения ранней секреции
1. Генетический дефект механизма восприятия

Патогенез СД 2 (продолжение)
Механизмы нарушения ранней секреции
2. Наследственное ↓ продукции
Патогенез СД 2 (продолжение)
Механизмы нарушения ранней секреции
2. Наследственное ↓ продукции

Характеристика СД 2 типа
1. Начало заболевания - в зрелом или пожилом
Характеристика СД 2 типа
1. Начало заболевания - в зрелом или пожилом

Лечение СД 2-го типа
1. Немедикаментозная терапия
Основа – постулат: «Диабет -
Лечение СД 2-го типа
1. Немедикаментозная терапия
Основа – постулат: «Диабет -

Лечение СД 2-го типа (продолжение)
2. Медикаментозная терапия
• Препараты, снижающие всасываемость глюкозы
Лечение СД 2-го типа (продолжение)
2. Медикаментозная терапия
• Препараты, снижающие всасываемость глюкозы

Хирургическое лечение СД 2 типа
1. Метаболическая хирургия:
- желудочное (гастрошунтирование) и
-
Хирургическое лечение СД 2 типа
1. Метаболическая хирургия:
- желудочное (гастрошунтирование) и
-
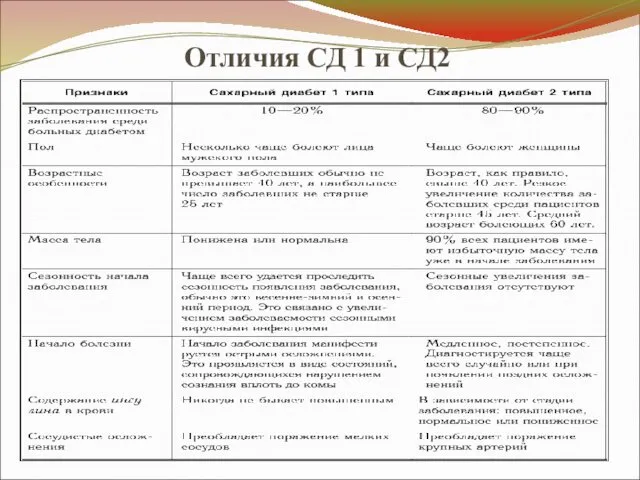
Отличия СД 1 и СД2
Отличия СД 1 и СД2

Гестационный СД
Возникает при беременности.
Может полностью исчезнуть после родов.
ГСД - нарушение
Гестационный СД
Возникает при беременности.
Может полностью исчезнуть после родов.
ГСД - нарушение

Гестационный СД
Влияет на:
• плод:
- внутриутробная гибель плода (из-за кетоза или поражения сосудов плаценты
Гестационный СД
Влияет на:
• плод:
- внутриутробная гибель плода (из-за кетоза или поражения сосудов плаценты

Другие специфические формы СД
1) СД при:
- органических поражениях поджелудочной железы (хронический
Другие специфические формы СД
1) СД при:
- органических поражениях поджелудочной железы (хронический

Другие специфические формы СД (продолжение)
2) СД, вызванный гиперпродукцией сахароповышающих гормонов (глюкагон,
Другие специфические формы СД (продолжение)
2) СД, вызванный гиперпродукцией сахароповышающих гормонов (глюкагон,

Другие специфические формы СД (продолжение)
3) СД, вызванный:
• химическими веществами (многие из
Другие специфические формы СД (продолжение)
3) СД, вызванный:
• химическими веществами (многие из

Другие специфические формы СД (продолжение)
4) Моногенные формы инсулинонезависимого СД - обусловлены
Другие специфические формы СД (продолжение)
4) Моногенные формы инсулинонезависимого СД - обусловлены
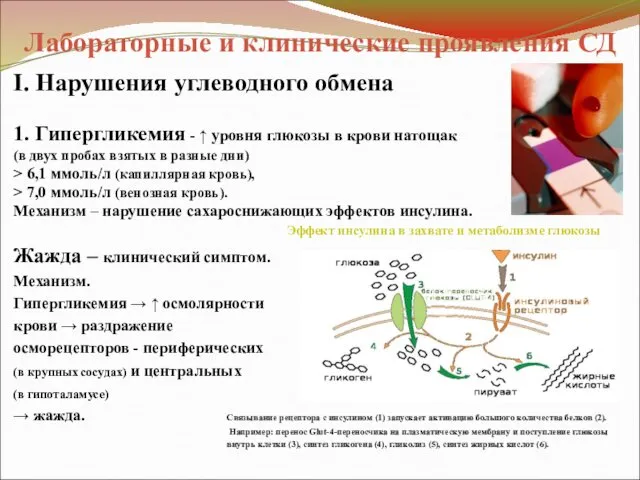
Лабораторные и клинические проявления СД
I. Нарушения углеводного обмена
1. Гипергликемия - ↑
Лабораторные и клинические проявления СД
I. Нарушения углеводного обмена
1. Гипергликемия - ↑

Лабораторные и клинические проявления СД (продолжение)
2. Глюкозурия – появление глюкозы в
Лабораторные и клинические проявления СД (продолжение)
2. Глюкозурия – появление глюкозы в

Лабораторные и клинические
проявления СД (продолжение)
3. Нарушение толерантности к глюкозе –
↓
Лабораторные и клинические
проявления СД (продолжение)
3. Нарушение толерантности к глюкозе –
↓
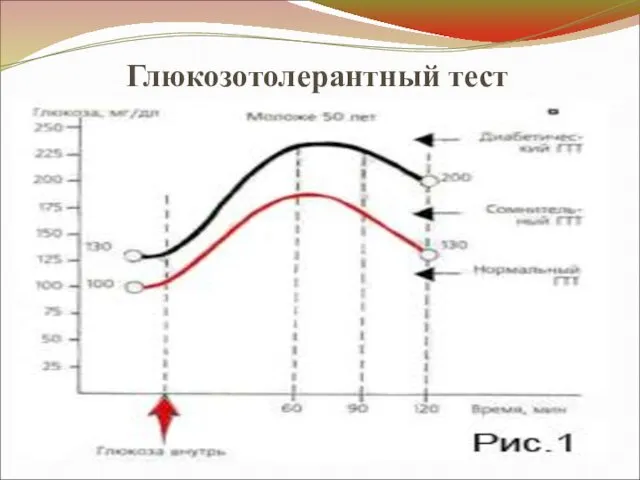
Глюкозотолерантный тест
Глюкозотолерантный тест

Другие критерии диагностики СД
4. ↑ содержания в крови гликозилированного (гликированного) Hb (HbА1:
Другие критерии диагностики СД
4. ↑ содержания в крови гликозилированного (гликированного) Hb (HbА1:

Диагностика гестационного СД
Заподозрить развитие у женщины СД беременных можно по анализу
Диагностика гестационного СД
Заподозрить развитие у женщины СД беременных можно по анализу

Нарушения липидного обмена
Лучше изучены для СД 1 типа.
Абсолютный дефицит инсулина нарушает
Нарушения липидного обмена
Лучше изучены для СД 1 типа.
Абсолютный дефицит инсулина нарушает

Нарушения липидного обмена (продолжение)
Расщепление ТГ формирует большой объем СЖК
→ выбрасываются
Нарушения липидного обмена (продолжение)
Расщепление ТГ формирует большой объем СЖК
→ выбрасываются

Нарушения липидного обмена (продолжение)
2. Большая часть окисляется до ацетил-КоА:
из-за нарушения утилизации
Нарушения липидного обмена (продолжение)
2. Большая часть окисляется до ацетил-КоА:
из-за нарушения утилизации

Нарушения белкового обмена
Инсулин – мощный анаболический гормон (стимулирует синтез нуклеиновых кислот,
Нарушения белкового обмена
Инсулин – мощный анаболический гормон (стимулирует синтез нуклеиновых кислот,
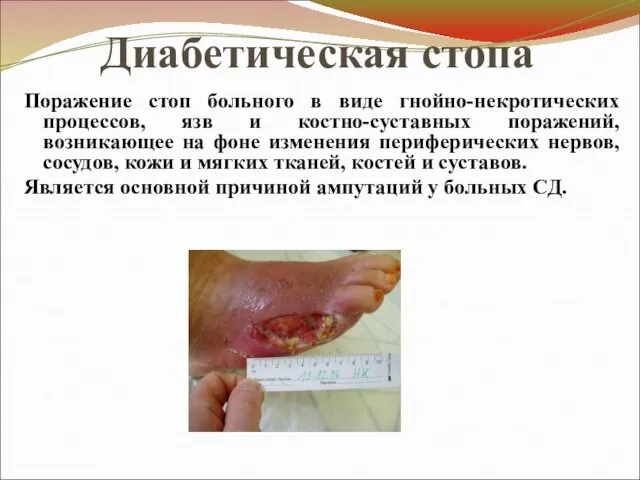
Диабетическая стопа
Поражение стоп больного в виде гнойно-некротических процессов, язв и
Диабетическая стопа
Поражение стоп больного в виде гнойно-некротических процессов, язв и

Водно-электролитный обмен
Экскреция кетоновых тел почками уменьшает содержание в организме связанных оснований,
Водно-электролитный обмен
Экскреция кетоновых тел почками уменьшает содержание в организме связанных оснований,

Классификация осложнений
I. Острые – кома:
• собственно диабетическая:
- кетоацидотическая,
- гиперосмолярная,
- лактацидотическая,
• гипогликемическая
II.
Классификация осложнений
I. Острые – кома:
• собственно диабетическая:
- кетоацидотическая,
- гиперосмолярная,
- лактацидотическая,
• гипогликемическая
II.

Кетоацидотическая кома
Обусловлена токсическим влиянием кетоновых тел на клетки ЦНС (блокируют выработку
Кетоацидотическая кома
Обусловлена токсическим влиянием кетоновых тел на клетки ЦНС (блокируют выработку

Гиперосмолярная кома
Встречается главным образом у пожилых больных с СД 2
Гиперосмолярная кома
Встречается главным образом у пожилых больных с СД 2

Лактацидотическая кома
Чаще возникает у больных старше 50 лет на фоне сердечно-сосудистой,
Лактацидотическая кома
Чаще возникает у больных старше 50 лет на фоне сердечно-сосудистой,

Гипогликемическая кома
Этиология
• передозировка инсулина или пероральных сахароснижающих препаратов,
• нарушение диеты
Гипогликемическая кома
Этиология
• передозировка инсулина или пероральных сахароснижающих препаратов,
• нарушение диеты

Микроангиопатии
М. – это комплекс специфических (характерны только для СД) патологических изменений
Микроангиопатии
М. – это комплекс специфических (характерны только для СД) патологических изменений

Микроангиопатии (продолжение)
Механизмы микроангиопатий
Главный патогенетический фактор - хроническая гипергликемия - индуцирует
Микроангиопатии (продолжение)
Механизмы микроангиопатий
Главный патогенетический фактор - хроническая гипергликемия - индуцирует

Механизмы микроангиопатий (продолжение)
• повышенное аутоокисление глюкозы
→ активация процессов ПОЛ
→
Механизмы микроангиопатий (продолжение)
• повышенное аутоокисление глюкозы
→ активация процессов ПОЛ
→

Диабетические макроангиопатии
МА - поражения артерий крупного и среднего калибра, с
Диабетические макроангиопатии
МА - поражения артерий крупного и среднего калибра, с

Макроангиопатии при СД (продолжение)
Диабетические МА (в соответствии с локализацией и клиническими
Макроангиопатии при СД (продолжение)
Диабетические МА (в соответствии с локализацией и клиническими

Макроангиопатии при СД (продолжение)
Дополнительно:
• ↓ ЛПВП + ↑ ЛПНП (нарушения липидного
Макроангиопатии при СД (продолжение)
Дополнительно:
• ↓ ЛПВП + ↑ ЛПНП (нарушения липидного

Профилактика осложнений
Контроль АД.
Преимущество отдаётся:
- метаболически нейтральным (ИАПФ, сартаны) и
- метаболически
Профилактика осложнений
Контроль АД.
Преимущество отдаётся:
- метаболически нейтральным (ИАПФ, сартаны) и
- метаболически
 Выделительная система. Строение и функции почек. Образование мочи
Выделительная система. Строение и функции почек. Образование мочи Туберкулез и его профилактика
Туберкулез и его профилактика Дифференциальная диагностика острого аппендицита с гинекологической и урологической патологией
Дифференциальная диагностика острого аппендицита с гинекологической и урологической патологией Аллергодерматозы у детей
Аллергодерматозы у детей Буллезный дерматоз. Пузырчатка
Буллезный дерматоз. Пузырчатка Анатомия грудной клетки
Анатомия грудной клетки Фитотерапия кожных заболеваний
Фитотерапия кожных заболеваний Тыныс жетіспеушілігі туралы жалпы түсінік
Тыныс жетіспеушілігі туралы жалпы түсінік Особенности психотерапевтической реабилитации пациентов с различными видами нехимических зависимостей
Особенности психотерапевтической реабилитации пациентов с различными видами нехимических зависимостей Психология личности
Психология личности Общий (гематологический) анализ крови
Общий (гематологический) анализ крови Гематология: лейкозы, геморрагические диатезы
Гематология: лейкозы, геморрагические диатезы Воля и волевые качества человека
Воля и волевые качества человека Емдік профилактикалық мекемелерде емдік дене шынықтыруды ұйымдастыру
Емдік профилактикалық мекемелерде емдік дене шынықтыруды ұйымдастыру Потребность пациента в нормальном дыхании
Потребность пациента в нормальном дыхании Сестринский персонал в программах профилактики ВИЧ. Лекция 1
Сестринский персонал в программах профилактики ВИЧ. Лекция 1 Вирусный гепатит D
Вирусный гепатит D Опухолевый рост
Опухолевый рост Systematization of grammar: "direct" and "indirect" speech. theme: Pathology of the form and structure of the teeth
Systematization of grammar: "direct" and "indirect" speech. theme: Pathology of the form and structure of the teeth Сучасний стан онкологічної служби в Україні. Організація онкологічної допомоги. Диспансеризація та облік онкологічних хворих
Сучасний стан онкологічної служби в Україні. Організація онкологічної допомоги. Диспансеризація та облік онкологічних хворих HIPEC при раке яичников
HIPEC при раке яичников Миокардит. Клинические признаки
Миокардит. Клинические признаки Исследование ликвора
Исследование ликвора Компенсаторно – приспособительные реакции
Компенсаторно – приспособительные реакции Поддерживающая терапия в период химиотерапии
Поддерживающая терапия в период химиотерапии Мариинская городская больница
Мариинская городская больница Возрастная анатомия, физиология и школьная гигиена
Возрастная анатомия, физиология и школьная гигиена Профилактическая медицина. Лекция № 1. История развития профилактики
Профилактическая медицина. Лекция № 1. История развития профилактики