-
Наследственная патология Моногенные синдромы

Содержание
- 2. Классификация моногенных синдромов Так как проявление моногенных болезней зависит от природы мутантного гена, существует их классификация
- 3. Аутосомно-доминантный тип наследования характеризуется следующими признаками: больные имеются в каждом поколении; больной ребенок у больных родителей;
- 4. Аутосомно-доминантный тип наследования Брахидактилия – короткопалость, которая выражается в укорочении фаланговых костей на пальцах. Существует целая
- 5. Брахидактилия Причиной заболевания являются гетерозиготные мутации в гене тирозин-киназного рецептора ROR2. Установлено, что этот ген экспрессируется
- 6. Робинов синдром Гомозиготные (доминантные и рецессивные) мутации в гене ROR2 обуславливают Робинов синдром, характеризующийся поражением конечностей
- 7. Аутосомно-доминантный тип наследования Ретинобластома — злокачественная опухоль эмбриональной сетчатки глаза. Встречается примерно у 1 новорожденного на
- 8. Ретинобластома если ребёнок наследует мутантный аллель гена Rb, то вторая мутация, происходящая уже в ретинобласте, ведёт
- 9. Аутосомно-доминантный тип наследования Нейрофиброматоз - тяжелая многосистемная болезнь. Популяционная частота - 1:3500 новорожденных. Ген картирован -
- 10. Нейрофиброматоз Чаще наблюдается нейрофиброматоз I типа (болезнь Реклингаузена). В этом случае имеются доминантные мутации в гене
- 11. Аутосомно-доминантный тип наследования Ахондроплазия (хондродистрофия) – диспропорциональная карликовость. У больных нарушаются рост и развитие хрящевой ткани
- 12. Ахондроплазия ахондроплазия вызывается мутацией в гене, кодирующем белок FGFR3 (рецептор 3 к фактору роста фибробластов). При
- 13. Аутосомно-доминантный тип наследования Синдром Марфана (архнодактилия) болезнь, причиной которой является мутация гена белка фибриллина (15q21). Популяционная
- 14. Синдром Марфана ?
- 15. Аутосомно-рецессивный тип наследования характеризуется следующими признаками: больные не в каждом поколении; больной ребенок (гомозигота) рождается у
- 16. Альбинизм врожденное отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза встречается в разных популяциях с
- 17. Альбинизм у представителей разных рас (из ru.wikipedia.org)
- 18. Типы альбинизма Глазокожный альбинизм 1 А - самая тяжелая форма альбинизма. Он появляется в результате миссенс,
- 19. Фенилкетонурия (ФКУ) встречается с частотой 1:6000 - 1:10 000. Вызвана мутацией гена, который отвечает за синтез
- 20. Фенилкетонурия Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает
- 21. Фенилкетонурия (из R.Lewis, 1994)
- 22. Алкаптонурия метаболическое заболевание, обусловленное мутацией гена АР (3q25-26), кодирующего синтез оксидазы гомогентизиновой кислоты. этот дефект приводит
- 23. Алкаптонурия Для больных характерны признаки поражения опорно-двигательного аппарата (артриты, остеоартрозы), внесуставных хрящевых структур и мягких тканей
- 24. Тирозинемия заболевание, связанное с дефицитом активности фумарилацетоацетатгидроксилазы. Ген локализован на 15-й хромосоме (15q23-q25). Мутации приводят к
- 25. Врожденные нарушения метаболизма фенилаланина и тирозина
- 26. Прогерия Больные прогерией часто имеют характерный внешний вид: низкий рост, относительно большая голова и уменьшенная лицевая
- 27. Прогерия характеризуется комплексом изменений кожи и внутренних органов, обусловленных преждевременным старением организма. Основными формами является детская
- 28. Детская прогерия Причина детской прогерии — мутации гена LMNA, кодирующего ламин А (1q22). Вероятный тип наследования:
- 29. Детская прогерия клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется рост ребенка, отмечаются атрофические
- 30. Детская прогерия Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство источников указывают возраст смерти
- 31. синдром Вернера Прогерия взрослых имеет аутосомно-рецессивный тип наследования (мутации в гене WRN, локализованном в хромосоме 8p12-p11.2).
- 32. ДНК-геликазы расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических процессов: репликация ДНК, транскрипция РНК
- 33. синдром Вернера Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост, гипогонадизм. Обычно на третьем
- 34. Синдром Блума Обусловлен мутациями в гене BLM, принадлежащем к генам ДНК-геликаз. Тип наследования – аутосомно-рецессивный (19q13.3).
- 35. Ксеродерма пигментная Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету (ультрафиолету), что проявляется в
- 36. Ксеродерма пигментная заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов. Существует семь комплементарных групп генов
- 37. Типы пигментной ксеродермы
- 38. Атаксия-телангиэктазия Синдром Луи-Бар: характерны прогрессивная мозжечковая атаксия с потерей клеток Пуркинье в мозжечке, телангиэктазия (расширение кровеносных
- 39. Локализация гена Atm (11q22.3), кодирующего киназу ATM, главный сенсор повреждения ДНК в клетке. Распознавая повреждение ДНК
- 40. Анемия Фанкони (Fanconi anemia) Развивается у детей в возрасте от 4 до 10 лет. Характеризуются аплазией
- 41. Анемия Фанкони Всего известно 7 генов, способных приводить к анемии Фанкони: FancA, FancB, FancC, FancD, FancE,
- 42. Анемия Фанкони Анемия Фанкони как и предыдущие заболевания (с. Блума, пигментная ксеродерма, атаксия-телангиэктазия) относятся к разряду
- 43. Роль теломер в старении Во многих клетках человека утрата способности клеток к делению связана с утратой
- 44. Общие причины синдромов преждевременного старения Нарушение свойств теломер, хроматина и клеточного ядра Нарушение репарации и репликации
- 45. Х-сцепленный рецессивный тип наследования характеризуется следующими признаками: больные появляются не в каждом поколении; больной ребенок рождается
- 46. Дальтонизм - частичная цветовая слепота, один из видов нарушения цветового зрения. Д. впервые описан в 1794
- 48. Гемофилия А - тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается с частотой 1:2500 новорожденных
- 49. Синдром Леша-Нихана (Lesch-Nyhan) обусловлен недостаточностью фермента гипоксантинфосфорибозилтрансферазы (ГФРТ), который катализирует присоединение свободных пуриновых оснований (гуанина и
- 50. Синдром Леша-Нихана Ген, кодирующий фермент гипоксантинфосфо-рибозилтрансферазу локализован в длином плече Х-хромосомы (Xq26-q27.2) Синдром встречается редко (1:300000
- 51. Миодистрофия Дюшенна - тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в плазме крови креатинфосфокиназы. Фенотипически
- 52. Х-сцепленный доминантный тип наследования сходен с аутосомно-доминантным, за исключением того, что мужчина передает этот признак только
- 54. Скачать презентацию

Классификация моногенных синдромов
Так как проявление моногенных болезней зависит от природы мутантного
Классификация моногенных синдромов
Так как проявление моногенных болезней зависит от природы мутантного

Аутосомно-доминантный тип наследования
характеризуется следующими признаками:
больные имеются в каждом
Аутосомно-доминантный тип наследования
характеризуется следующими признаками:
больные имеются в каждом
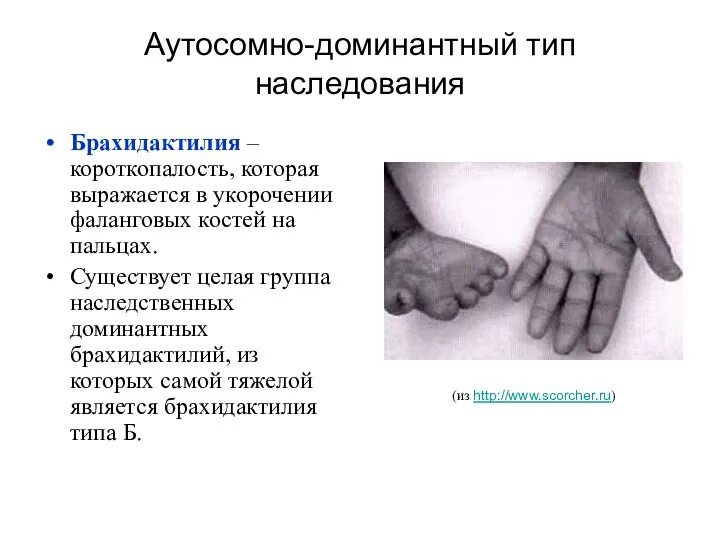
Аутосомно-доминантный тип наследования
Брахидактилия – короткопалость, которая выражается в укорочении фаланговых костей
Аутосомно-доминантный тип наследования
Брахидактилия – короткопалость, которая выражается в укорочении фаланговых костей

Брахидактилия
Причиной заболевания являются гетерозиготные мутации в гене тирозин-киназного рецептора ROR2.
Установлено, что
Брахидактилия
Причиной заболевания являются гетерозиготные мутации в гене тирозин-киназного рецептора ROR2.
Установлено, что
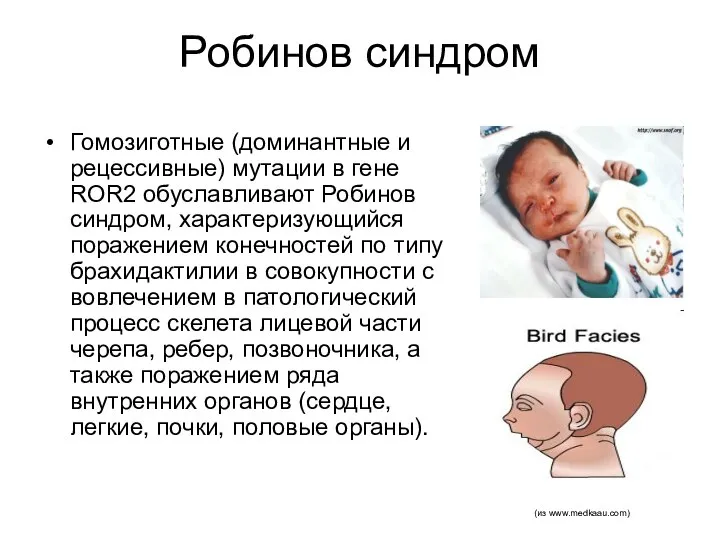
Робинов синдром
Гомозиготные (доминантные и рецессивные) мутации в гене ROR2 обуславливают Робинов
Робинов синдром
Гомозиготные (доминантные и рецессивные) мутации в гене ROR2 обуславливают Робинов
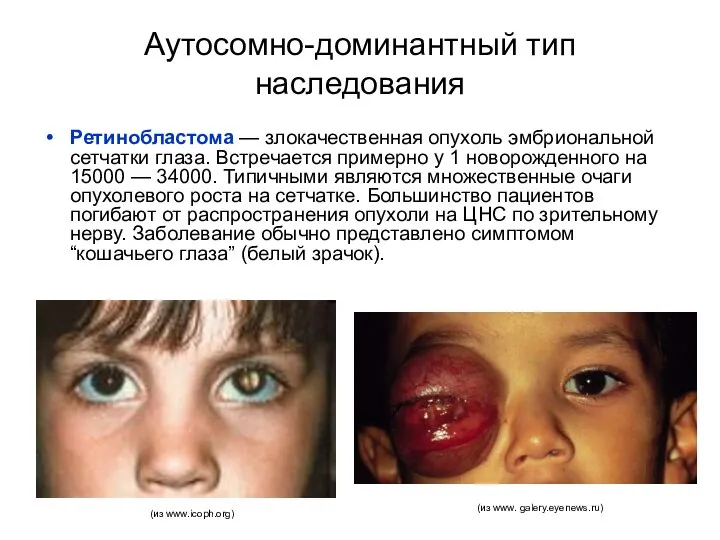
Аутосомно-доминантный тип наследования
Ретинобластома — злокачественная опухоль эмбриональной сетчатки глаза. Встречается примерно
Аутосомно-доминантный тип наследования
Ретинобластома — злокачественная опухоль эмбриональной сетчатки глаза. Встречается примерно

Ретинобластома
если ребёнок наследует мутантный аллель гена Rb, то вторая мутация, происходящая
Ретинобластома
если ребёнок наследует мутантный аллель гена Rb, то вторая мутация, происходящая
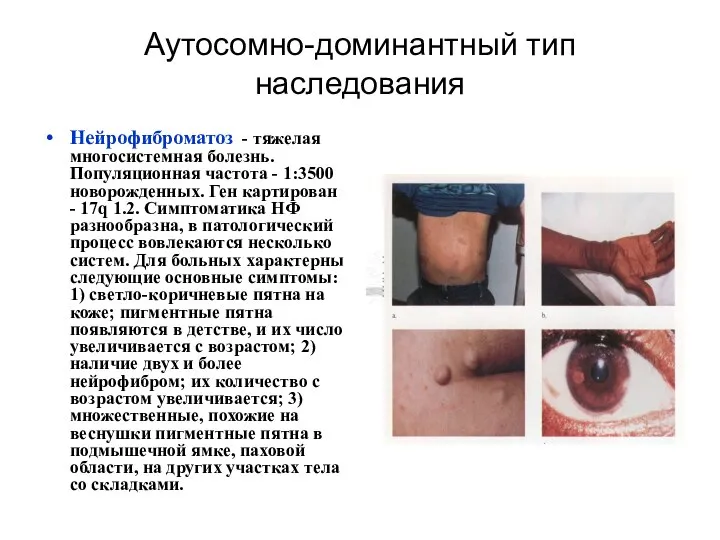
Аутосомно-доминантный тип наследования
Нейрофиброматоз - тяжелая многосистемная болезнь. Популяционная частота - 1:3500
Аутосомно-доминантный тип наследования
Нейрофиброматоз - тяжелая многосистемная болезнь. Популяционная частота - 1:3500

Нейрофиброматоз
Чаще наблюдается нейрофиброматоз I типа (болезнь Реклингаузена). В этом случае имеются
Нейрофиброматоз
Чаще наблюдается нейрофиброматоз I типа (болезнь Реклингаузена). В этом случае имеются
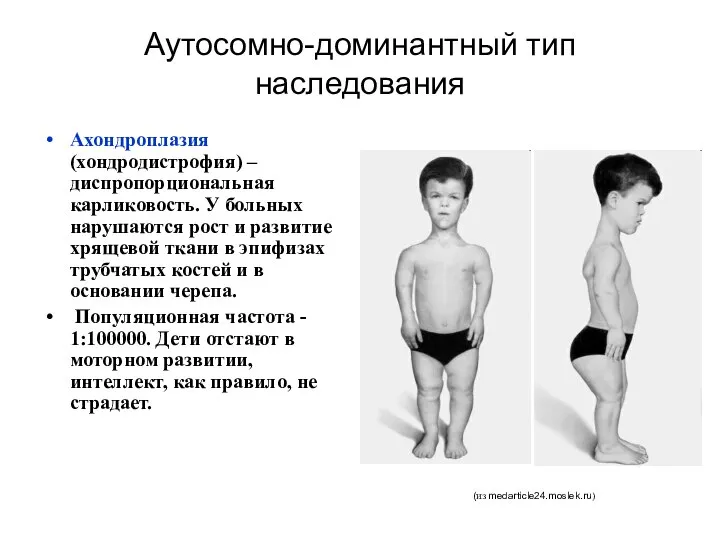
Аутосомно-доминантный тип наследования
Ахондроплазия (хондродистрофия) – диспропорциональная карликовость. У больных нарушаются рост
Аутосомно-доминантный тип наследования
Ахондроплазия (хондродистрофия) – диспропорциональная карликовость. У больных нарушаются рост

Ахондроплазия
ахондроплазия вызывается мутацией в гене, кодирующем белок FGFR3 (рецептор 3 к
Ахондроплазия
ахондроплазия вызывается мутацией в гене, кодирующем белок FGFR3 (рецептор 3 к
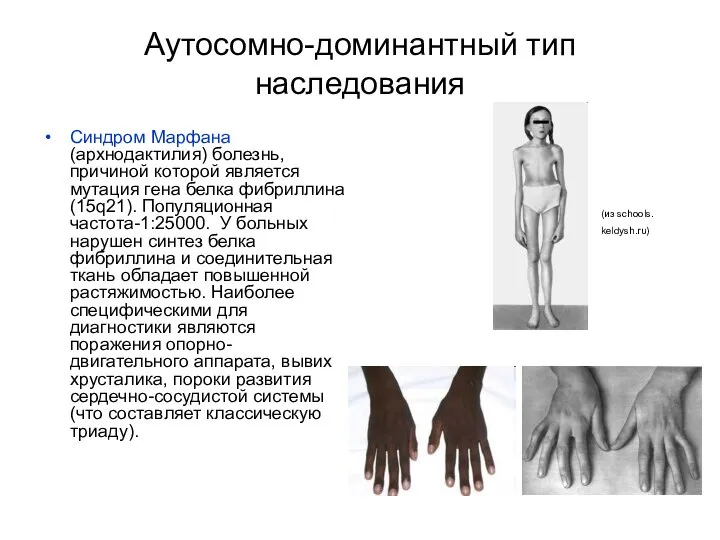
Аутосомно-доминантный тип наследования
Синдром Марфана (архнодактилия) болезнь, причиной которой является мутация гена
Аутосомно-доминантный тип наследования
Синдром Марфана (архнодактилия) болезнь, причиной которой является мутация гена

Синдром Марфана ?
Синдром Марфана ?

Аутосомно-рецессивный тип наследования
характеризуется следующими признаками:
больные не в каждом
Аутосомно-рецессивный тип наследования
характеризуется следующими признаками:
больные не в каждом

Альбинизм
врожденное отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза встречается в
Альбинизм
врожденное отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза встречается в

Альбинизм у представителей разных рас
(из ru.wikipedia.org)
Альбинизм у представителей разных рас
(из ru.wikipedia.org)

Типы альбинизма
Глазокожный альбинизм 1 А - самая тяжелая форма альбинизма. Он
Типы альбинизма
Глазокожный альбинизм 1 А - самая тяжелая форма альбинизма. Он
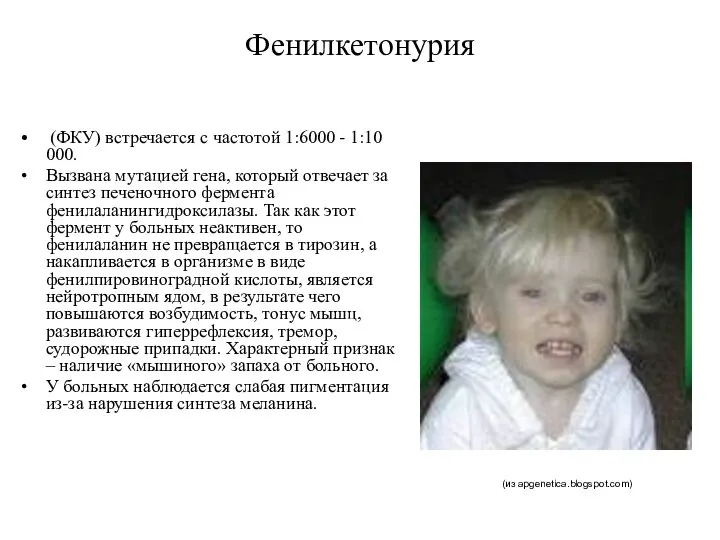
Фенилкетонурия
(ФКУ) встречается с частотой 1:6000 - 1:10 000.
Вызвана мутацией
Фенилкетонурия
(ФКУ) встречается с частотой 1:6000 - 1:10 000.
Вызвана мутацией

Фенилкетонурия
Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и
Фенилкетонурия
Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и
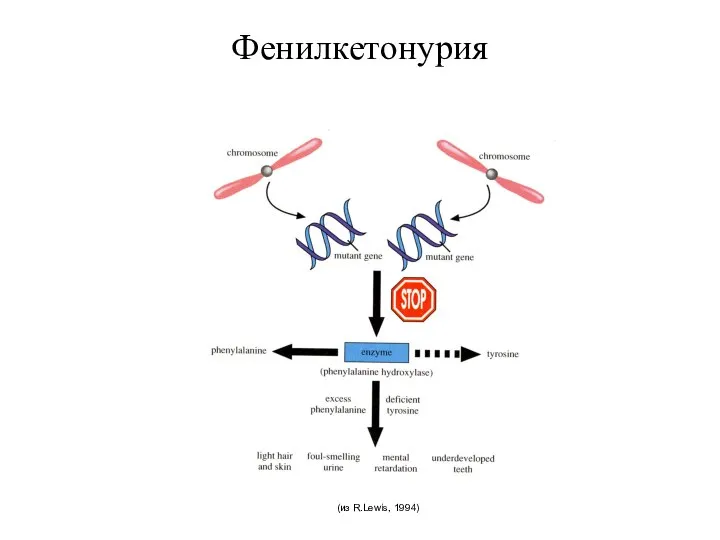
Фенилкетонурия
(из R.Lewis, 1994)
Фенилкетонурия
(из R.Lewis, 1994)

Алкаптонурия
метаболическое заболевание, обусловленное мутацией гена АР (3q25-26), кодирующего синтез оксидазы гомогентизиновой
Алкаптонурия
метаболическое заболевание, обусловленное мутацией гена АР (3q25-26), кодирующего синтез оксидазы гомогентизиновой
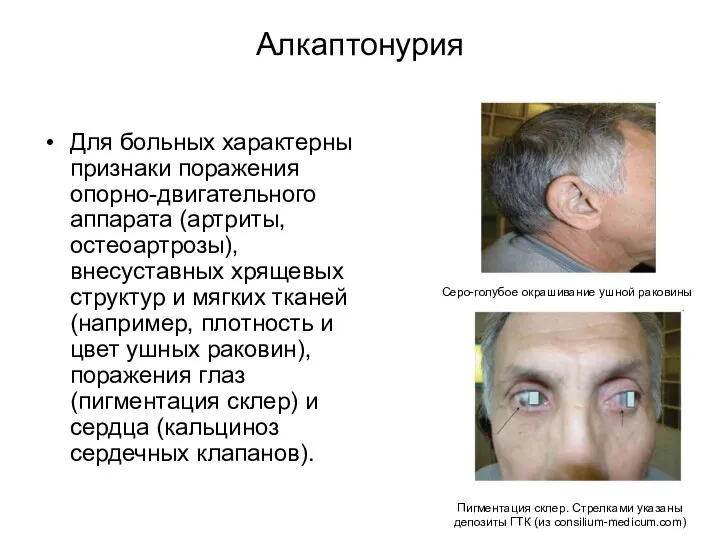
Алкаптонурия
Для больных характерны признаки поражения опорно-двигательного аппарата (артриты, остеоартрозы), внесуставных хрящевых
Алкаптонурия
Для больных характерны признаки поражения опорно-двигательного аппарата (артриты, остеоартрозы), внесуставных хрящевых

Тирозинемия
заболевание, связанное с дефицитом активности фумарилацетоацетатгидроксилазы. Ген локализован на 15-й хромосоме
Тирозинемия
заболевание, связанное с дефицитом активности фумарилацетоацетатгидроксилазы. Ген локализован на 15-й хромосоме
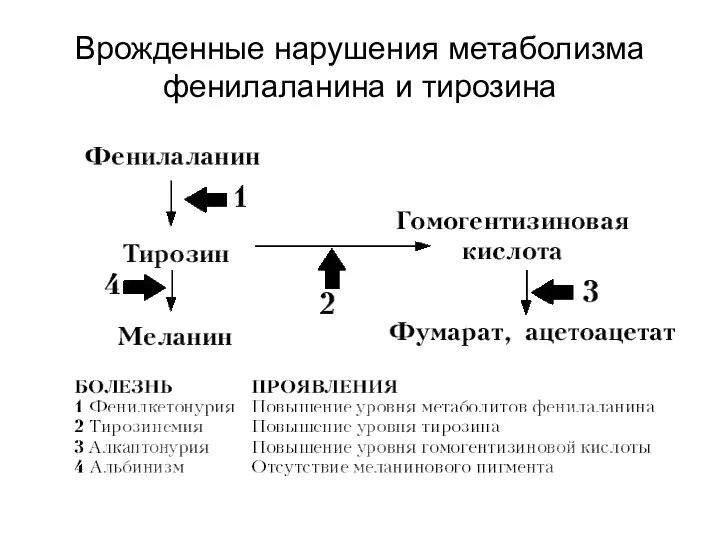
Врожденные нарушения метаболизма фенилаланина и тирозина
Врожденные нарушения метаболизма фенилаланина и тирозина
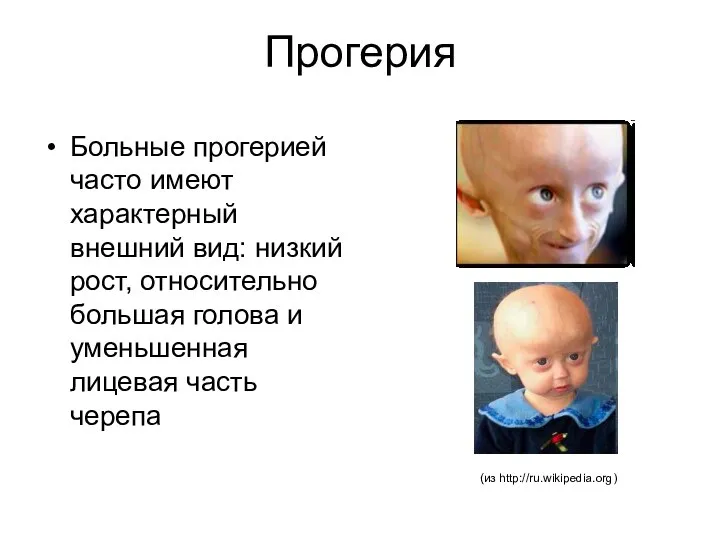
Прогерия
Больные прогерией часто имеют характерный внешний вид: низкий рост, относительно большая
Прогерия
Больные прогерией часто имеют характерный внешний вид: низкий рост, относительно большая

Прогерия
характеризуется комплексом изменений кожи и внутренних органов, обусловленных преждевременным старением организма.
Прогерия
характеризуется комплексом изменений кожи и внутренних органов, обусловленных преждевременным старением организма.

Детская прогерия
Причина детской прогерии — мутации гена LMNA, кодирующего ламин А
Детская прогерия
Причина детской прогерии — мутации гена LMNA, кодирующего ламин А

Детская прогерия
клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется
Детская прогерия
клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется

Детская прогерия
Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство
Детская прогерия
Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство
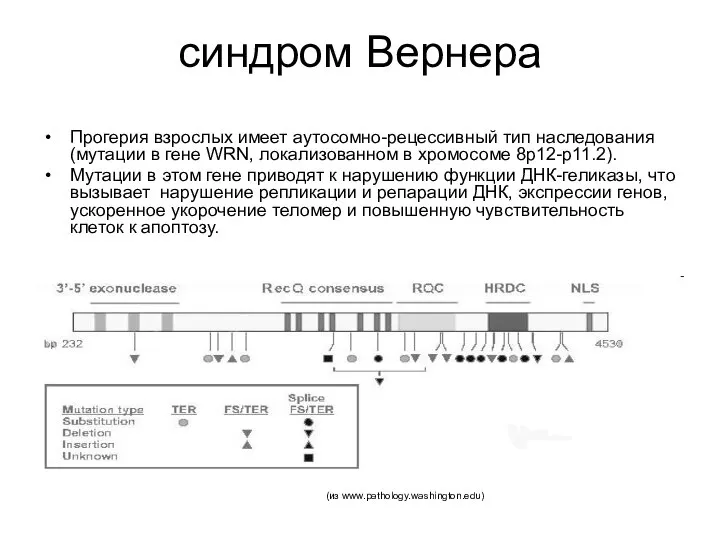
синдром Вернера
Прогерия взрослых имеет аутосомно-рецессивный тип наследования (мутации в гене WRN,
синдром Вернера
Прогерия взрослых имеет аутосомно-рецессивный тип наследования (мутации в гене WRN,
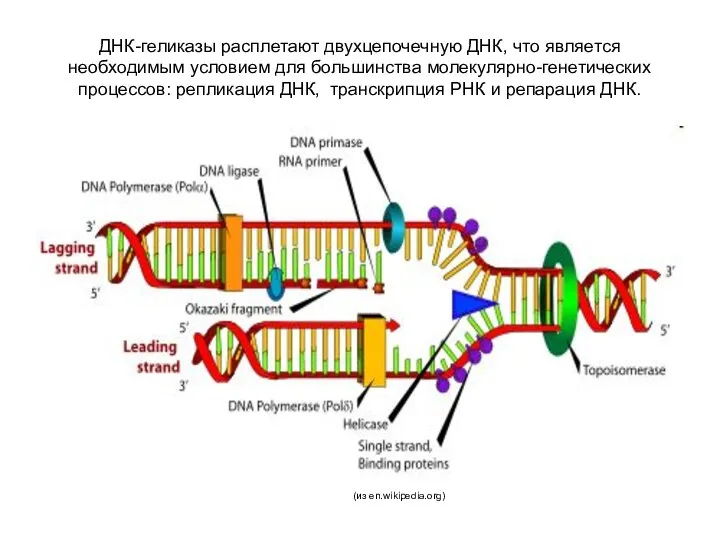
ДНК-геликазы расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических
ДНК-геликазы расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических

синдром Вернера
Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост,
синдром Вернера
Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост,

Синдром Блума
Обусловлен мутациями в гене BLM, принадлежащем к генам ДНК-геликаз. Тип
Синдром Блума
Обусловлен мутациями в гене BLM, принадлежащем к генам ДНК-геликаз. Тип
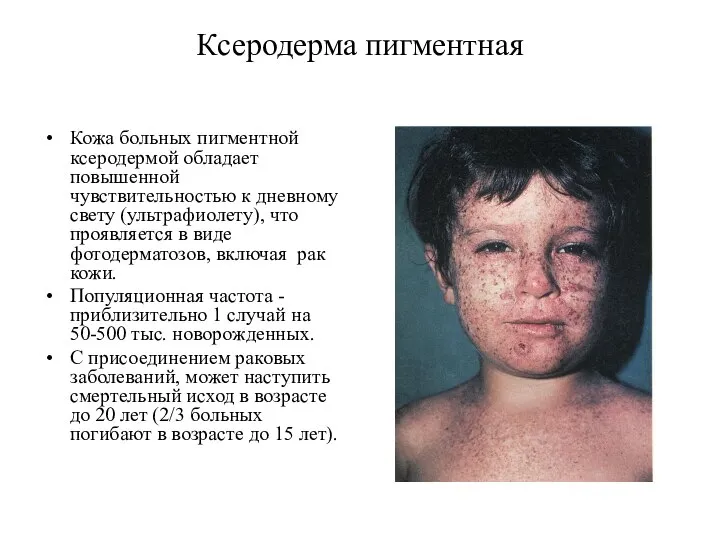
Ксеродерма пигментная
Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету
Ксеродерма пигментная
Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету

Ксеродерма пигментная
заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов.
Существует
Ксеродерма пигментная
заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов.
Существует
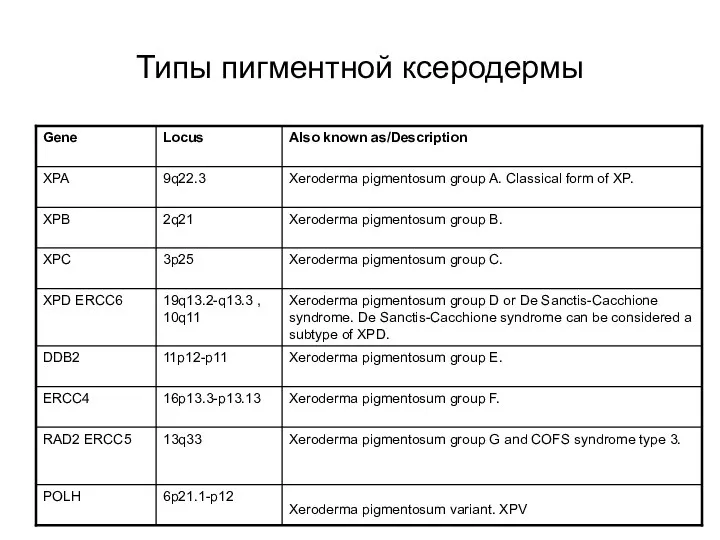
Типы пигментной ксеродермы
Типы пигментной ксеродермы
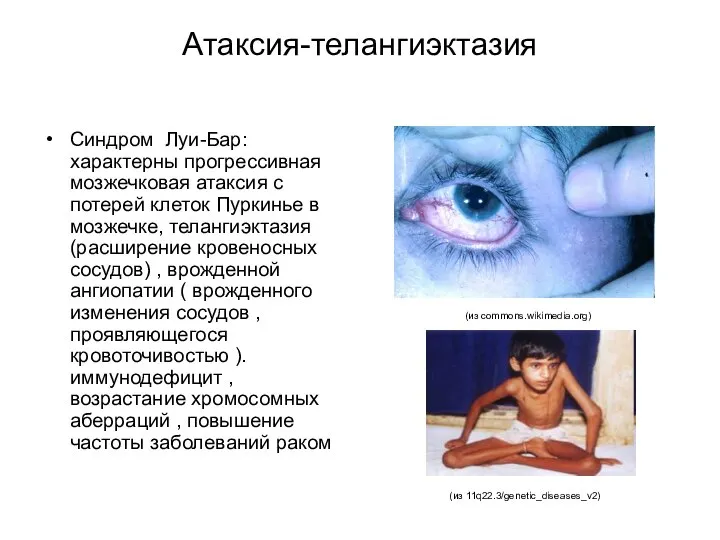
Атаксия-телангиэктазия
Синдром Луи-Бар: характерны прогрессивная мозжечковая атаксия с потерей клеток Пуркинье в
Атаксия-телангиэктазия
Синдром Луи-Бар: характерны прогрессивная мозжечковая атаксия с потерей клеток Пуркинье в
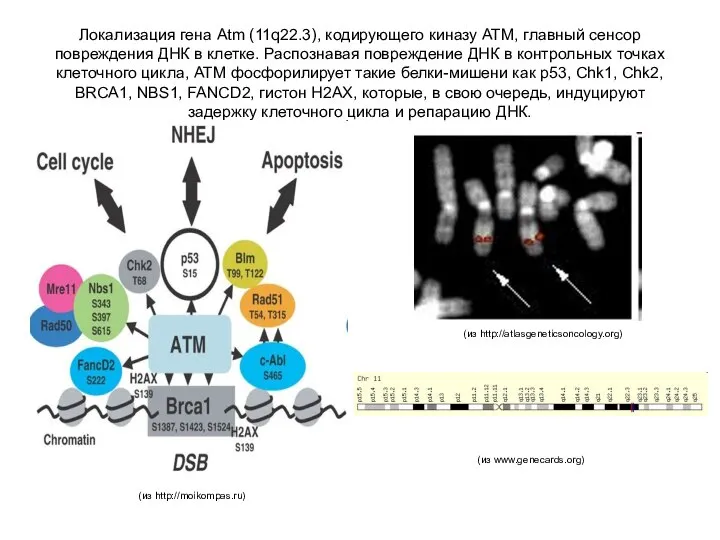
Локализация гена Atm (11q22.3), кодирующего киназу ATM, главный сенсор повреждения ДНК
Локализация гена Atm (11q22.3), кодирующего киназу ATM, главный сенсор повреждения ДНК
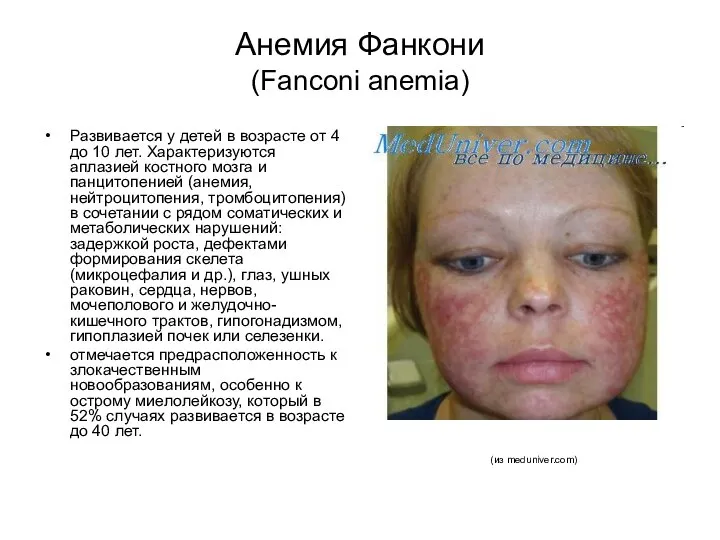
Анемия Фанкони
(Fanconi anemia)
Развивается у детей в возрасте от 4 до
Анемия Фанкони
(Fanconi anemia)
Развивается у детей в возрасте от 4 до
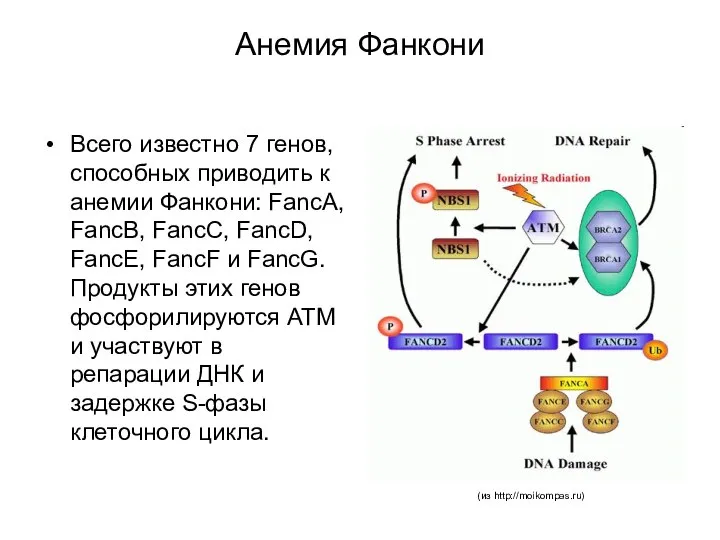
Анемия Фанкони
Всего известно 7 генов, способных приводить к анемии Фанкони: FancA,
Анемия Фанкони
Всего известно 7 генов, способных приводить к анемии Фанкони: FancA,
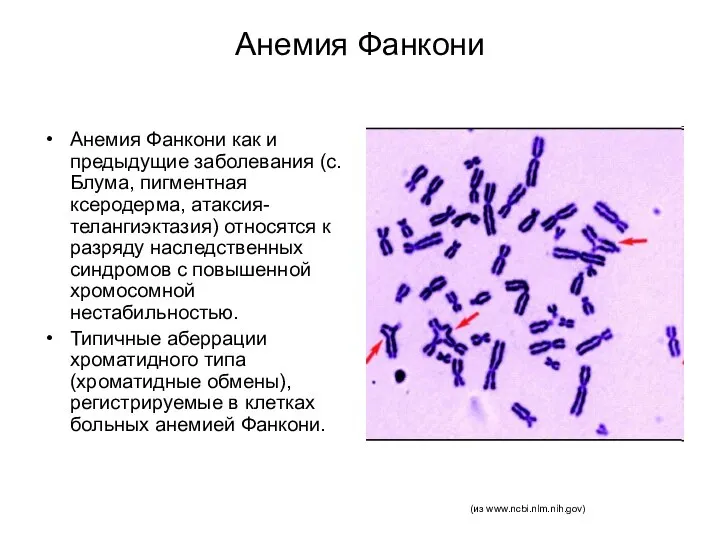
Анемия Фанкони
Анемия Фанкони как и предыдущие заболевания (с. Блума, пигментная ксеродерма,
Анемия Фанкони
Анемия Фанкони как и предыдущие заболевания (с. Блума, пигментная ксеродерма,
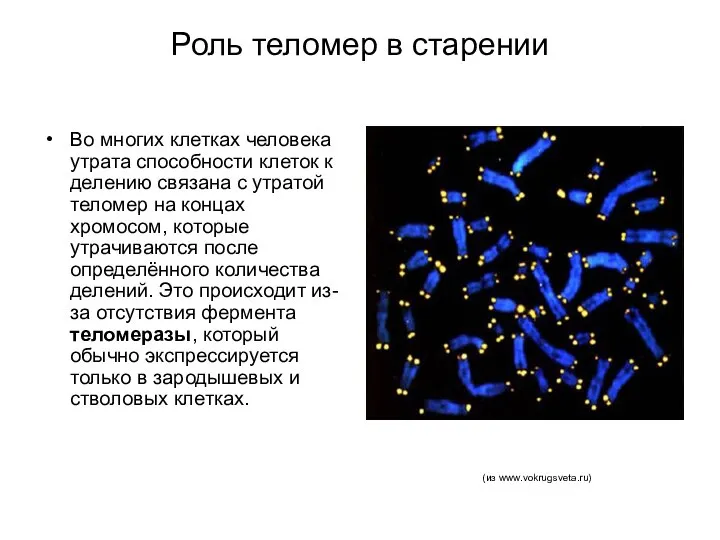
Роль теломер в старении
Во многих клетках человека утрата способности клеток к
Роль теломер в старении
Во многих клетках человека утрата способности клеток к

Общие причины синдромов преждевременного старения
Нарушение свойств теломер, хроматина и клеточного ядра
Общие причины синдромов преждевременного старения
Нарушение свойств теломер, хроматина и клеточного ядра

Х-сцепленный рецессивный тип наследования
характеризуется следующими признаками:
больные появляются не в
Х-сцепленный рецессивный тип наследования
характеризуется следующими признаками:
больные появляются не в

Дальтонизм
- частичная цветовая слепота, один из видов нарушения цветового зрения. Д.
Дальтонизм
- частичная цветовая слепота, один из видов нарушения цветового зрения. Д.


Гемофилия А
- тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается
Гемофилия А
- тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается

Синдром Леша-Нихана (Lesch-Nyhan)
обусловлен недостаточностью фермента гипоксантинфосфорибозилтрансферазы (ГФРТ), который катализирует присоединение
Синдром Леша-Нихана (Lesch-Nyhan)
обусловлен недостаточностью фермента гипоксантинфосфорибозилтрансферазы (ГФРТ), который катализирует присоединение

Синдром Леша-Нихана
Ген, кодирующий фермент гипоксантинфосфо-рибозилтрансферазу локализован в длином плече Х-хромосомы (Xq26-q27.2)
Синдром
Синдром Леша-Нихана
Ген, кодирующий фермент гипоксантинфосфо-рибозилтрансферазу локализован в длином плече Х-хромосомы (Xq26-q27.2)
Синдром
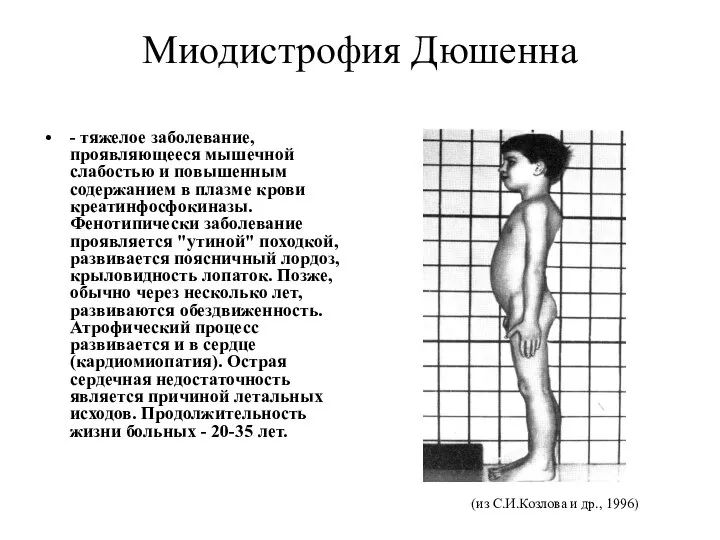
Миодистрофия Дюшенна
- тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в
Миодистрофия Дюшенна
- тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в

Х-сцепленный доминантный тип наследования
сходен с аутосомно-доминантным, за исключением того, что
Х-сцепленный доминантный тип наследования
сходен с аутосомно-доминантным, за исключением того, что
 Діяльність спортивного клубу у ВНЗ
Діяльність спортивного клубу у ВНЗ Halloween is celebrated on the 31-st of October
Halloween is celebrated on the 31-st of October Заболевания сосудов
Заболевания сосудов Кластеризация поисковых запросов. Разработка структуры сайта
Кластеризация поисковых запросов. Разработка структуры сайта Культура Київської Русі (ІХ-ХІІІ ст.)
Культура Київської Русі (ІХ-ХІІІ ст.) Нестабилизированный заход на посадку
Нестабилизированный заход на посадку Презентация по алгебре Теорема синусов
Презентация по алгебре Теорема синусов  Проект поселения будущего
Проект поселения будущего 6 секретов няни, открывшей счёт в Швейцарском банке: Тонкости профессии НЯНЯ Автор Саламатова Александра
6 секретов няни, открывшей счёт в Швейцарском банке: Тонкости профессии НЯНЯ Автор Саламатова Александра Презентация на тему "Пищеварение в кишечнике" - скачать презентации по Медицине
Презентация на тему "Пищеварение в кишечнике" - скачать презентации по Медицине 1 Базовые конструкции языка программирования. - презентация
1 Базовые конструкции языка программирования. - презентация Презентация Рынок угля
Презентация Рынок угля  Физическая культура в профессиональной подготовке студентов и социокультурное развитие личности студента
Физическая культура в профессиональной подготовке студентов и социокультурное развитие личности студента Вибрация
Вибрация Причіпні пожежні мотопомпи. Експлуатація мотопомп
Причіпні пожежні мотопомпи. Експлуатація мотопомп Тварина - живий організм
Тварина - живий організм Elections in USA
Elections in USA Инструктор по тхэквон-до
Инструктор по тхэквон-до Динамика распространения волн в упругой среде . Ур-ие Даламбера
Динамика распространения волн в упругой среде . Ур-ие Даламбера  Поняття, предмет та система кримінології. Історія розвитку кримінології
Поняття, предмет та система кримінології. Історія розвитку кримінології Профилактика и реабилитация вчера, сегодня, завтра Доц. каф. ТПСД, к.м.н. Сагадеева Е.М.
Профилактика и реабилитация вчера, сегодня, завтра Доц. каф. ТПСД, к.м.н. Сагадеева Е.М. Термины по теме «Право собственности»
Термины по теме «Право собственности»  Встреча студентов СПБГЭУ с Генеральным консулом США в Санкт-Петербурге, Томасом Лири
Встреча студентов СПБГЭУ с Генеральным консулом США в Санкт-Петербурге, Томасом Лири Рабочая профессия слесарь-электрик по ремонту электрооборудования
Рабочая профессия слесарь-электрик по ремонту электрооборудования Настольный теннис. Основные правила игры
Настольный теннис. Основные правила игры Скиния собрания
Скиния собрания Монтаж сборных строительных конструкций промышленного здания
Монтаж сборных строительных конструкций промышленного здания Вся жизнь Японии - искусство - презентация для начальной школы
Вся жизнь Японии - искусство - презентация для начальной школы