- Наследственная изменчивость. Мутационная изменчивость. Генные мутации

Содержание
- 2. План занятия. 1. Определение мутационной изменчивости. 2. Основные положения мутационной теории. 3. Классификация мутаций: генные, геномные,
- 3. ИЗМЕНЧИВОСТЬ ФЕНОТИПИЧЕСКАЯ (модификационная, ненаследственная) КОМБИНАТИВНАЯ ГЕНОТИПИЧЕСКАЯ (наследственная) МУТАЦИОННАЯ ГЕННЫЕ МУТАЦИИ ГЕНОМНЫЕ МУТАЦИИ ХРОМОСОМНЫЕ АБЕРРАЦИИ
- 4. 1. Определение мутационной изменчивости. Наследственная (генотипическая) изменчивость проявляется в изменении генотипа особи, поэтому передается при половом
- 5. Мутационная изменчивость - обусловленная возникновением мутаций. Мутация — это устойчивое и ненаправленное изменение в геноме.
- 6. 2. Основные положения мутационной теории. Мутационна теория почти одновременно зародилась в умах голландца Хуго де Фриза
- 7. Основные положения мутационной теории. 1. Мутации внезапны, как дискретные изменения признаков. 2. Новые формы устойчивы. 3.
- 8. Мутационная теория Исследования Х. Де Фриза проводились на различных видах Ослинника, которые в ходе эксперимента не
- 9. В. Иогансен - строгое доказательство возникновения мутаций. - Эксперименты на самоопыляющихся линиях фасоли и ячменя: были
- 10. Основные положения мутационной теории. 1. Мутации внезапны, как дискретные изменения признаков. 2. Новые формы устойчивы. 3.
- 11. Противоречие между мутационной и эволюционной теориями. Ч. Дарвин: ЕСТЕСТВЕННЫЙ ОТБОР. Коржинский и де Фриз: МУТАЦИОННАЯ ИЗМЕНЧИВОСТЬ
- 12. «Кошмар Дженкина» В июне 1867 года в журнале «North British Review» вышла в свет статья Дженкина
- 13. Флеминг Дженкин: Представим себе белого человека, потерпевшего кораблекрушение на острове, населённом неграми… Наш выживший герой, возможно,
- 14. Ошибка Дженкина заключалась в том, что признаки, закрепляемые отбором, не уменьшаются при скрещивании, а передаются в
- 15. Закон гомологических рядов в наследственной изменчивости Н.И. Вавилова (1920). Крупнейшее обобщение работ по изучению изменчивости в
- 16. Мутации: - Мутации возникают постоянно на протяжении всего онтогенеза человека. Чем на более раннем этапе развития
- 17. Универсальность мутационной изменчивости. Мутации SARS-CoV-2: 1. Британская мутация B.1.1.7 2. Южноафриканская мутация B.1.351 3. Бразильская мутация
- 18. Универсальность мутационной изменчивости.
- 19. Мутационный процесс СПОНТАННЫЙ- протекает под влиянием естественных факторов ИНДУЦИРОВАННЫЙ- - целенаправленное воздействии на клетку Частота спонтанного
- 20. 3. Классификация мутаций
- 21. Мутации по типу действия на организм: - нейтральные; - вредные; - полезные. Современные генетики считают: -
- 22. Вредность и полезность мутаций — понятия относительные.
- 23. Мутации: - Большинство мутаций являются рецессивными; - Рецессивный характер мутантных аллелей позволяет им длительное время сохраняться
- 24. Мутации по характеру действия: - морфологические, - физиологические, - биохимические.
- 25. Морфологические мутации Брахидактилия - Изменяют формирование органов и ростовые процессы у животных и растений. Карликовость растений
- 26. Физиологические мутации Хлорофильные мутации растений - Обычно понижают жизнеспособность особей, среди них много летальных и полулетальных
- 27. Биохимические мутации Фенилкетонурия - отсутствие фермента синтезирующего тирозин из фенилаланина, в результате чего фенилаланин накапливается в
- 28. Мутации по месту возникновения: ГЕНЕРАТИВНЫЕ: - Возникают в половых клетках - Передаются потомству при половом размножении;
- 29. Мутагенез — процесс возникновения мутаций.
- 30. Мутации по характеру изменения генома:
- 31. ГЕННЫЕ МУТАЦИИ -дупликации —повторение участка гена, - вставки— появление в последовательности лишней пары нуклеотидов, - делеции
- 32. ГЕННЫЕ МУТАЦИИ
- 33. Причины генных заболеваний Центральная догма молекулярной биологии (Ф.Крик, 1968) В результате мутации гена на молекулярном уровне
- 34. Генные мутации могут быть связаны с нарушением обмена углеводов, липидов, стероидов, пуринов и пиримидинов, билирубина, металлов
- 35. Фенилкетонурия Причина: Генный дефект - отсутствие или недостаточная активность фермента фенилаланингидроксидазы, что приводит к нарушению обмена
- 36. Фенилкетонурия
- 37. Алкаптонурия Причина: Нарушение обмена фенилаланина и тирозина и экскреция с мочой гомогентизиновои кислоты. Тип наследования: Аутосомно-рецессивный.
- 38. Алкаптонурия
- 39. Катаракта Причина: Генная мутация, обусловливающая врожденное помутнение хрусталика. Тип наследования: Аутосомно-доминантный, реже аутосомно-рецессивный. Клиника: Снижение зрения
- 40. Гемофилия Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой мутацией гена, локализованного в
- 41. Анемия Фанкони (наследственная апластическая) Причина: возникает при наличии дефекта в кластере белков, отвечающих за репарацию ДНК
- 42. Галактоземия Причина: Происходит накопление в крови больного галактозы, что приводит к поражению многих органов: печени, нервной
- 43. Болезнь Гоше Причина: Причиной болезни Гоше является дефект гена глюкоцереброзидазы. Глюкоцереброзидаза - это фермент, который помогает
- 44. Рахит, витамин D-зависимый Причина: В крови гипокальцемия, повышение содержания витамина D в 10-100 раз. Тип наследования:
- 45. Альбинизм Причина: Врожденное отсутствие или инактивация энзима тирозиназы в эпителиальных клетках, в связи, с чем нарушается
- 46. Синдром Марфана Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена. Тип наследования: Аутосомно-доминантный. Клиника: Высокий
- 47. Мукополисахаридоз Причина: Мукополисахаридозы представлены целой группой наследственных заболеваний соединительной ткани. Для них характерно нарушение в организме
- 48. Серповидно-клеточная анемия Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный гемолиз и распад эритроцитов,
- 49. Нейрофиброматоз (болезнь Реклингхаузена) Причина: У больных в печени, почках и слизистой кишечника накапливается большое количество гликогена.
- 50. Синдром Беквита - Видемана Причина: Отмечается гипогликемия, сочетающаяся с рядом соматических изменений, что может являться причиной
- 51. Синдром Вильямса, «лицо Эльфа». Причина: У детей повышенный уровень кальция в сыворотке крови. Тип наследования: Аутосомно-доминантный.
- 52. Синдром Энгельмана Причина: инактивация генов области g11-13 хромосомы 15 материнского происхождения. Тип наследования: Аутосомно-доминантный. Клиника: Болезнь
- 53. Ахондроплазия Причина: Из-за сниженной способности больных иметь потомство в 80-95% случаев это заболевание связано с заново
- 54. Полидактилия Причина: Генная мутация. Тип наследования: Аутосомно-доминантный. Клиника: Увеличение количества (до 8-12) пальцев на кистях и
- 56. Скачать презентацию

План занятия.
1. Определение мутационной изменчивости.
2. Основные положения мутационной теории.
3. Классификация мутаций:
План занятия.
1. Определение мутационной изменчивости.
2. Основные положения мутационной теории.
3. Классификация мутаций:
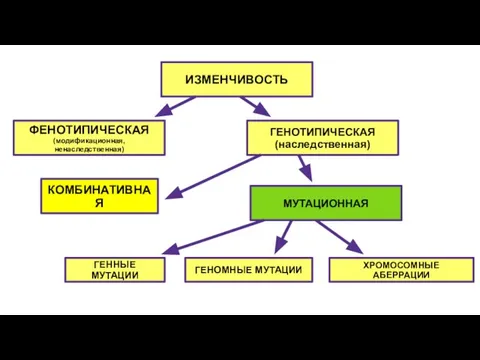
ИЗМЕНЧИВОСТЬ
ФЕНОТИПИЧЕСКАЯ
(модификационная, ненаследственная)
КОМБИНАТИВНАЯ
ГЕНОТИПИЧЕСКАЯ
(наследственная)
МУТАЦИОННАЯ
ГЕННЫЕ МУТАЦИИ
ГЕНОМНЫЕ МУТАЦИИ
ХРОМОСОМНЫЕ АБЕРРАЦИИ
ИЗМЕНЧИВОСТЬ
ФЕНОТИПИЧЕСКАЯ
(модификационная, ненаследственная)
КОМБИНАТИВНАЯ
ГЕНОТИПИЧЕСКАЯ
(наследственная)
МУТАЦИОННАЯ
ГЕННЫЕ МУТАЦИИ
ГЕНОМНЫЕ МУТАЦИИ
ХРОМОСОМНЫЕ АБЕРРАЦИИ

1. Определение мутационной изменчивости.
Наследственная (генотипическая) изменчивость проявляется в изменении генотипа особи,
1. Определение мутационной изменчивости.
Наследственная (генотипическая) изменчивость проявляется в изменении генотипа особи,

Мутационная изменчивость - обусловленная возникновением мутаций.
Мутация — это устойчивое и
Мутационная изменчивость - обусловленная возникновением мутаций.
Мутация — это устойчивое и

2. Основные положения мутационной теории.
Мутационна теория почти одновременно зародилась в умах
2. Основные положения мутационной теории.
Мутационна теория почти одновременно зародилась в умах

Основные положения мутационной теории.
1. Мутации внезапны, как дискретные изменения признаков.
2.
Основные положения мутационной теории.
1. Мутации внезапны, как дискретные изменения признаков.
2.

Мутационная теория
Исследования Х. Де Фриза проводились на различных видах Ослинника, которые
Мутационная теория
Исследования Х. Де Фриза проводились на различных видах Ослинника, которые

В. Иогансен - строгое доказательство возникновения мутаций.
- Эксперименты на самоопыляющихся линиях
В. Иогансен - строгое доказательство возникновения мутаций.
- Эксперименты на самоопыляющихся линиях

Основные положения мутационной теории.
1. Мутации внезапны, как дискретные изменения признаков.
2.
Основные положения мутационной теории.
1. Мутации внезапны, как дискретные изменения признаков.
2.

Противоречие между мутационной и эволюционной теориями.
Ч. Дарвин: ЕСТЕСТВЕННЫЙ ОТБОР.
Коржинский и де
Противоречие между мутационной и эволюционной теориями.
Ч. Дарвин: ЕСТЕСТВЕННЫЙ ОТБОР.
Коржинский и де

«Кошмар Дженкина»
В июне 1867 года в журнале «North British Review» вышла
«Кошмар Дженкина»
В июне 1867 года в журнале «North British Review» вышла

Флеминг Дженкин:
Представим себе белого человека, потерпевшего кораблекрушение на острове, населённом неграми…
Флеминг Дженкин:
Представим себе белого человека, потерпевшего кораблекрушение на острове, населённом неграми…
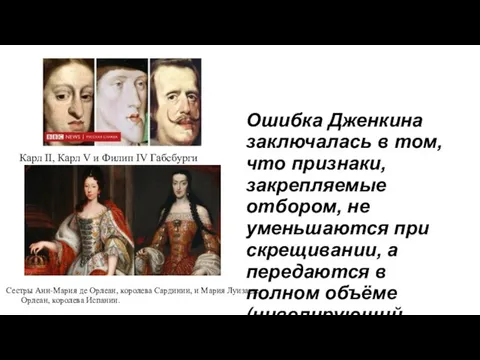
Ошибка Дженкина заключалась в том, что признаки, закрепляемые отбором, не уменьшаются
Ошибка Дженкина заключалась в том, что признаки, закрепляемые отбором, не уменьшаются

Закон гомологических рядов в наследственной изменчивости Н.И. Вавилова (1920).
Крупнейшее обобщение работ
Закон гомологических рядов в наследственной изменчивости Н.И. Вавилова (1920).
Крупнейшее обобщение работ
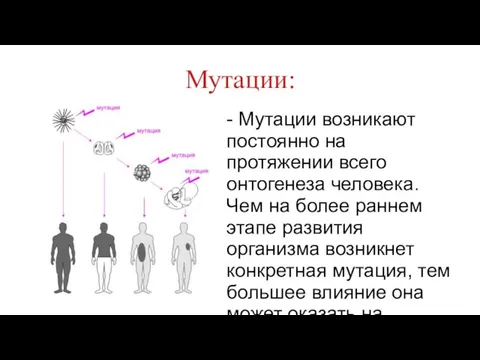
Мутации:
- Мутации возникают постоянно на протяжении всего онтогенеза человека. Чем на
Мутации:
- Мутации возникают постоянно на протяжении всего онтогенеза человека. Чем на
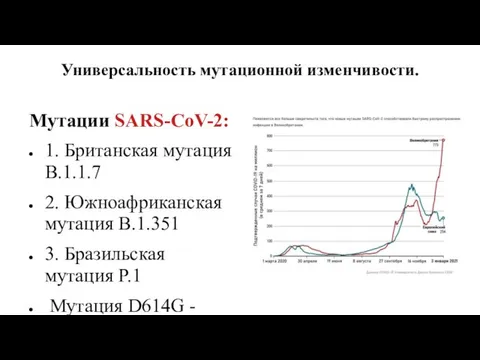
Универсальность мутационной изменчивости.
Мутации SARS-CoV-2:
1. Британская мутация B.1.1.7
2. Южноафриканская мутация B.1.351
3. Бразильская
Универсальность мутационной изменчивости.
Мутации SARS-CoV-2:
1. Британская мутация B.1.1.7
2. Южноафриканская мутация B.1.351
3. Бразильская

Универсальность мутационной изменчивости.
Универсальность мутационной изменчивости.

Мутационный процесс
СПОНТАННЫЙ-
протекает под влиянием естественных факторов
ИНДУЦИРОВАННЫЙ-
- целенаправленное воздействии на
Мутационный процесс
СПОНТАННЫЙ-
протекает под влиянием естественных факторов
ИНДУЦИРОВАННЫЙ-
- целенаправленное воздействии на

3. Классификация мутаций
3. Классификация мутаций

Мутации по типу действия на организм:
- нейтральные;
- вредные;
- полезные.
Современные генетики считают:
-
Мутации по типу действия на организм:
- нейтральные;
- вредные;
- полезные.
Современные генетики считают:
-
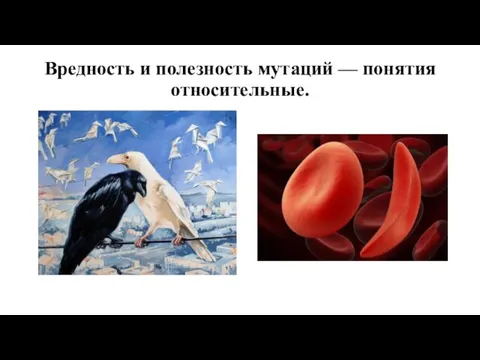
Вредность и полезность мутаций — понятия относительные.
Вредность и полезность мутаций — понятия относительные.

Мутации:
- Большинство мутаций являются рецессивными;
- Рецессивный характер мутантных аллелей позволяет им
Мутации:
- Большинство мутаций являются рецессивными;
- Рецессивный характер мутантных аллелей позволяет им

Мутации по характеру действия:
- морфологические,
- физиологические,
- биохимические.
Мутации по характеру действия:
- морфологические,
- физиологические,
- биохимические.

Морфологические мутации
Брахидактилия
- Изменяют формирование органов и ростовые процессы у животных
Морфологические мутации
Брахидактилия
- Изменяют формирование органов и ростовые процессы у животных

Физиологические мутации
Хлорофильные мутации растений
- Обычно понижают жизнеспособность особей, среди них много
Физиологические мутации
Хлорофильные мутации растений
- Обычно понижают жизнеспособность особей, среди них много

Биохимические мутации
Фенилкетонурия - отсутствие фермента синтезирующего тирозин из фенилаланина, в результате
Биохимические мутации
Фенилкетонурия - отсутствие фермента синтезирующего тирозин из фенилаланина, в результате

Мутации по месту возникновения:
ГЕНЕРАТИВНЫЕ:
- Возникают в половых клетках
- Передаются потомству при
Мутации по месту возникновения:
ГЕНЕРАТИВНЫЕ:
- Возникают в половых клетках
- Передаются потомству при
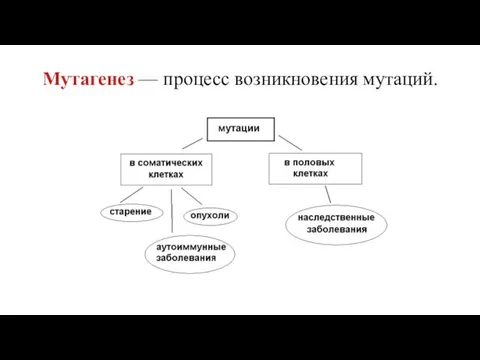
Мутагенез — процесс возникновения мутаций.
Мутагенез — процесс возникновения мутаций.

Мутации по характеру изменения генома:
Мутации по характеру изменения генома:

ГЕННЫЕ МУТАЦИИ
-дупликации —повторение участка гена,
- вставки— появление в последовательности лишней пары
ГЕННЫЕ МУТАЦИИ
-дупликации —повторение участка гена,
- вставки— появление в последовательности лишней пары

ГЕННЫЕ МУТАЦИИ
ГЕННЫЕ МУТАЦИИ

Причины генных заболеваний
Центральная догма молекулярной биологии (Ф.Крик, 1968)
В результате мутации гена
Причины генных заболеваний
Центральная догма молекулярной биологии (Ф.Крик, 1968)
В результате мутации гена

Генные мутации могут быть связаны с нарушением обмена углеводов, липидов, стероидов,
Генные мутации могут быть связаны с нарушением обмена углеводов, липидов, стероидов,

Фенилкетонурия
Причина: Генный дефект - отсутствие или недостаточная активность фермента фенилаланингидроксидазы, что
Фенилкетонурия
Причина: Генный дефект - отсутствие или недостаточная активность фермента фенилаланингидроксидазы, что

Фенилкетонурия
Фенилкетонурия

Алкаптонурия
Причина: Нарушение обмена фенилаланина и тирозина и экскреция с мочой гомогентизиновои
Алкаптонурия
Причина: Нарушение обмена фенилаланина и тирозина и экскреция с мочой гомогентизиновои
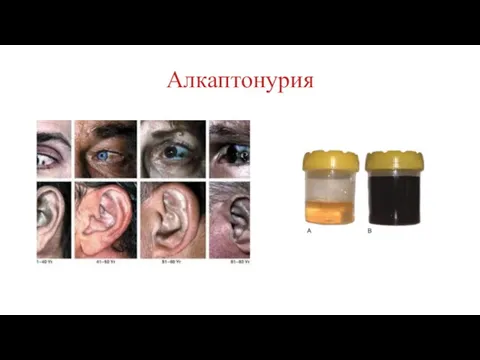
Алкаптонурия
Алкаптонурия
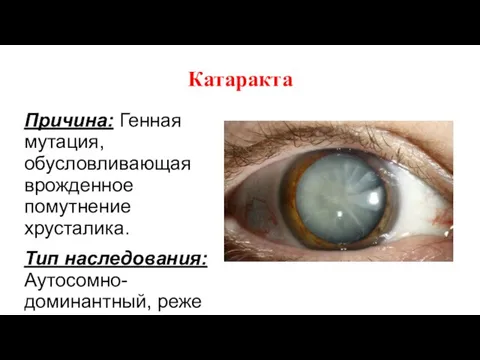
Катаракта
Причина: Генная мутация, обусловливающая врожденное помутнение хрусталика.
Тип наследования: Аутосомно-доминантный, реже аутосомно-рецессивный.
Катаракта
Причина: Генная мутация, обусловливающая врожденное помутнение хрусталика.
Тип наследования: Аутосомно-доминантный, реже аутосомно-рецессивный.
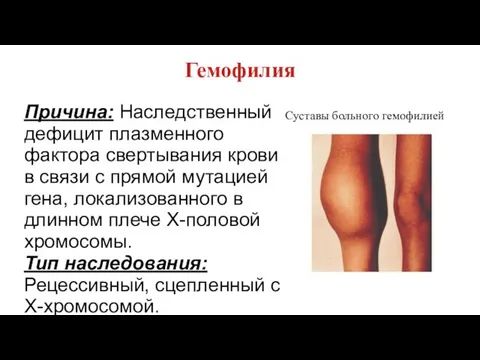
Гемофилия
Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой
Гемофилия
Причина: Наследственный дефицит плазменного фактора свертывания крови в связи с прямой

Анемия Фанкони (наследственная апластическая)
Причина: возникает при наличии дефекта в кластере белков,
Анемия Фанкони (наследственная апластическая)
Причина: возникает при наличии дефекта в кластере белков,
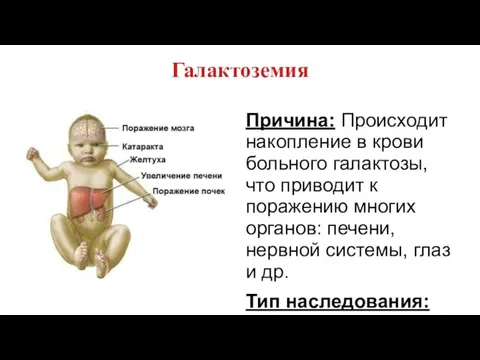
Галактоземия
Причина: Происходит накопление в крови больного галактозы, что приводит к поражению
Галактоземия
Причина: Происходит накопление в крови больного галактозы, что приводит к поражению

Болезнь Гоше
Причина: Причиной болезни Гоше является дефект гена глюкоцереброзидазы. Глюкоцереброзидаза -
Болезнь Гоше
Причина: Причиной болезни Гоше является дефект гена глюкоцереброзидазы. Глюкоцереброзидаза -

Рахит, витамин D-зависимый
Причина: В крови гипокальцемия, повышение содержания витамина D в
Рахит, витамин D-зависимый
Причина: В крови гипокальцемия, повышение содержания витамина D в

Альбинизм
Причина: Врожденное отсутствие или инактивация энзима тирозиназы в эпителиальных клетках, в
Альбинизм
Причина: Врожденное отсутствие или инактивация энзима тирозиназы в эпителиальных клетках, в
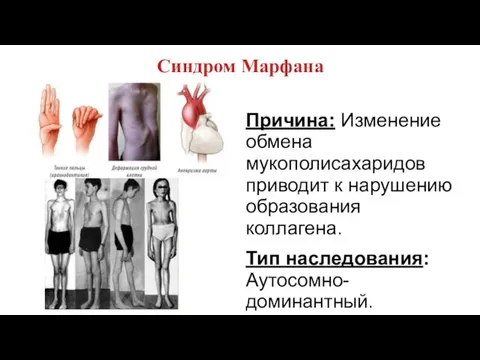
Синдром Марфана
Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена.
Тип наследования:
Синдром Марфана
Причина: Изменение обмена мукополисахаридов приводит к нарушению образования коллагена.
Тип наследования:

Мукополисахаридоз
Причина: Мукополисахаридозы представлены целой группой наследственных заболеваний соединительной ткани. Для них
Мукополисахаридоз
Причина: Мукополисахаридозы представлены целой группой наследственных заболеваний соединительной ткани. Для них

Серповидно-клеточная анемия
Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный
Серповидно-клеточная анемия
Причина: Генные мутации полипептидной цепочки гемоглобина; в результате наблюдается преждевременный
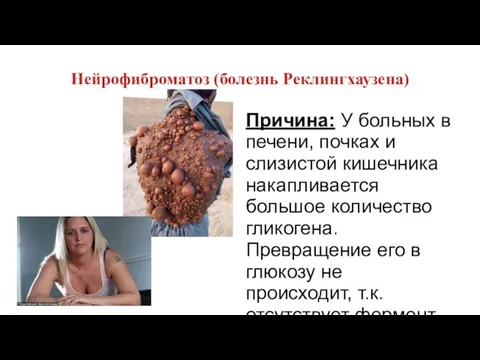
Нейрофиброматоз (болезнь Реклингхаузена)
Причина: У больных в печени, почках и слизистой кишечника
Нейрофиброматоз (болезнь Реклингхаузена)
Причина: У больных в печени, почках и слизистой кишечника
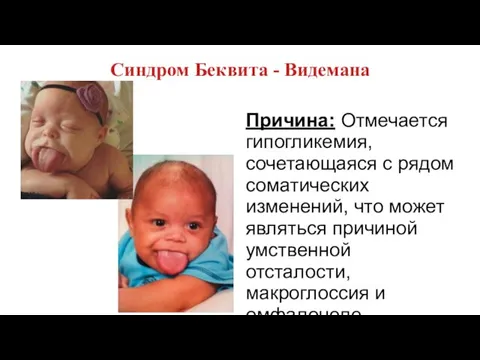
Синдром Беквита - Видемана
Причина: Отмечается гипогликемия, сочетающаяся с рядом соматических изменений,
Синдром Беквита - Видемана
Причина: Отмечается гипогликемия, сочетающаяся с рядом соматических изменений,

Синдром Вильямса, «лицо Эльфа».
Причина: У детей повышенный уровень кальция в
Синдром Вильямса, «лицо Эльфа».
Причина: У детей повышенный уровень кальция в

Синдром Энгельмана
Причина: инактивация генов области g11-13 хромосомы 15 материнского происхождения.
Тип наследования:
Синдром Энгельмана
Причина: инактивация генов области g11-13 хромосомы 15 материнского происхождения.
Тип наследования:

Ахондроплазия
Причина: Из-за сниженной способности больных иметь потомство в 80-95% случаев это
Ахондроплазия
Причина: Из-за сниженной способности больных иметь потомство в 80-95% случаев это

Полидактилия
Причина: Генная мутация.
Тип наследования: Аутосомно-доминантный.
Клиника: Увеличение количества (до 8-12) пальцев на
Полидактилия
Причина: Генная мутация.
Тип наследования: Аутосомно-доминантный.
Клиника: Увеличение количества (до 8-12) пальцев на
 Давай спасём бездомных животных вместе
Давай спасём бездомных животных вместе Презентация на тему Экологические кризисы и экологические катастрофы
Презентация на тему Экологические кризисы и экологические катастрофы Презентация Слуховой анализатор
Презентация Слуховой анализатор Презентация на тему "Метаболизм. Фотосинтез" - скачать презентации по Биологии
Презентация на тему "Метаболизм. Фотосинтез" - скачать презентации по Биологии Эволюция систем органов. Онтофилогенетическая обусловленность пороков развития
Эволюция систем органов. Онтофилогенетическая обусловленность пороков развития Функционирование белков
Функционирование белков Кровеносная система
Кровеносная система Краниометрическая программа. Краниометрические размеры. (Тема 3)
Краниометрическая программа. Краниометрические размеры. (Тема 3) Пищеварительная система. Остальные системы жизнедеятельности человека
Пищеварительная система. Остальные системы жизнедеятельности человека Урок на тему: «Ткани» биология 8 класс. Автор урока учитель биологии высшей категории МОУ СОШ № 43 г. Пензы. Петрунина Т.П.
Урок на тему: «Ткани» биология 8 класс. Автор урока учитель биологии высшей категории МОУ СОШ № 43 г. Пензы. Петрунина Т.П.  Идеальный рацион питания для зимующих птиц Кузбасса Битинёва Александра 8а класс МОУ СОШ № 5
Идеальный рацион питания для зимующих птиц Кузбасса Битинёва Александра 8а класс МОУ СОШ № 5 Палеонтология. История жизни на земле от бактерий до динозавров
Палеонтология. История жизни на земле от бактерий до динозавров Порфирины. Производные порфина. (Лекция 14)
Порфирины. Производные порфина. (Лекция 14) Бег лошади
Бег лошади Индуцированный мутагенез
Индуцированный мутагенез Презентация на тему "Геральдика и бонистика в преподавании биологии" - скачать презентации по Биологии
Презентация на тему "Геральдика и бонистика в преподавании биологии" - скачать презентации по Биологии Презентация на тему "Краткая история развития зоологии" - скачать презентации по Биологии
Презентация на тему "Краткая история развития зоологии" - скачать презентации по Биологии Предмет испытания и соревнования собак
Предмет испытания и соревнования собак Биоэлектрические явления в возбудимых тканях. Мембранные потенциалы: потенциал покоя, потенциал действия
Биоэлектрические явления в возбудимых тканях. Мембранные потенциалы: потенциал покоя, потенциал действия Десять самых красивых животных
Десять самых красивых животных Организмы, способные жить при крайних значениях температуры
Организмы, способные жить при крайних значениях температуры Генная и клеточная инженерия. Презентация по биологии
Генная и клеточная инженерия. Презентация по биологии Значение воздуха для жизни на Земле
Значение воздуха для жизни на Земле Корисні Бактерії (6 клас)
Корисні Бактерії (6 клас) Тема: Історичний розвиток і різноманітність органічного світу Єра динозаврів Виконала: Горовенко Надія
Тема: Історичний розвиток і різноманітність органічного світу Єра динозаврів Виконала: Горовенко Надія  Зеленые водоросли
Зеленые водоросли Синичкин день
Синичкин день Следы на снегу изучение и охрана диких зверей зимой
Следы на снегу изучение и охрана диких зверей зимой