- Генные и хромосомные болезни

Содержание
- 2. Вопросы 1. Общая характеристика моногенных болезней, классификация 2. Патогенез развития наследственных болезней обмена 3. Основные направления
- 3. Моногенные заболевания Распространенность- 1-2% среди новорожденных детей Описано более 4000 нозологических форм моногенных наследственных болезней, из
- 4. Моногенные заболевания Моногенные болезни - это группа наследственных заболеваний, в основе которых лежат генные мутации, возникшие
- 5. Мутации Мутация - любое изменение последовательности ДНК Мутационая теория де Фриза (1901-1903): 1. Мутации возникают скачкообразно,
- 6. Моногенные заболевания Виды генных мутаций 1. Со сдвигом рамки считывания 2. Мутации, обусловленные заменой азотистых оснований
- 7. Моногенные заболевания Виды генных мутаций По характеру изменений в молекуле ДНК: Со сдвигом рамки считывания –
- 8. Мутация сдвига рамки считывания Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ ЦЦА ЦГГ ТЦГ ЦАГ АТА
- 10. Моногенные заболевания Виды генных мутаций Мутации, обусловленные заменой азотистых оснований - транзиции -замена одного пурина на
- 11. Миссенс- мутации Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ ЦЦА ЦГГ ТЦГ ЦАГ АТА Нормальная мРНК
- 13. Моногенные заболевания Если в результате мутаций этой группы образуется терминирующий кодон (стоп кодон)–УАГ, УГА, УАА (и-РНК),
- 14. Нонсенс-мутации Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ ЦЦА ЦГГ ТЦГ ЦАГ АТА Нормальная мРНК ГГУ
- 16. Моногенные заболевания Генетическая гетерогенность - различия в генетической природе одного заболевания. Клинический полиморфизм – различия в
- 17. Источники генетической гетерогенности : 1) полилокусность (локус – это место расположения гена на хромосоме) А В
- 18. Источники генетической гетерогенности 2) полиаллелизм – явление, когда различные мутации одного локуса (гена) затрагивают одну и
- 19. Миодистрофия Дюшенна-Беккера / DMD Наследственное нервно-мышечное заболевание, характеризующееся прогрессирующими дегенеративными изменениями в поперечнополосатой мускулатуре. Молекулярный механизм
- 20. Клинически выделяют две формы миодистрофии: Миодистрофию Дюшена - мутация в гене (локусе) приводит к полному прекращению
- 22. Фенилкетонурия (ФКУ / PKU) Врожденное заболевание, вызванное нарушением перехода фенилаланина в тирозин, приводящее к задержке психического
- 23. Метаболические пути тирозина и фенилаланина в организме человека
- 25. Больной с фенилкетонурией, слабая пигментация кожи, волос, радужной оболочки глаз, умеренная степень олигофрении (Ивар Фелинг, 1934г.)
- 27. Факторы, обусловливающие клинический полиморфизм : генетическими факторами (полиаллелизм), модифицирующим влиянием средовых факторов, сочетанием этих двух групп
- 28. Моногенные заболевания Функции белков ферментативная, структурная транспортная.
- 29. Принципы классификации моногенных заболеваний Генетический Клинический Патогенетический
- 30. Моногенные заболевания наследуются по законам Менделя Болезни с аутосомно-доминантным типом наследования Болезни с аутосомно-рецессивный типом наследования
- 31. Классификация моногенных наследственных болезней: наследственные болезни обмена веществ ( нарушается работа белков-ферментов, а также могут быть
- 32. Клиническая классификация Заболевания: Нервные Нервно-мышечные Кожные Глазные Опорно-двигательного аппарата Эндокринные Крови Сердечно-сосудистой системы Мочеполовой системы Желудочно-кишечного
- 33. Наследственные болезни обмена наследственные дефекты обмена аминокислот (фенилкетонурия, гистидинемия, алкаптонурия, гомоцистинурия). наследственные дефекты обмена углеводов (галактоземия,
- 34. Наследственные болезни обмена наследственные дефекты биосинтеза кортикостероидов: адреногенитальный синдром, гипоальдостеронизм и др; наследственные дефекты порфиринового и
- 35. Ферментопатии — наследственные заболевания, обусловленные нарушением синтеза белков ферментов
- 36. Патогенез развития наследственных болезней обмена воздействие мутантного аллеля выработка патологического первичного продукта цепь последующих биохимических процессов
- 37. Варианты патологических эффектов мутантных генов 1. Мутантный ген приводит к синтезу избыточного количества продукта 2. Мутантный
- 38. Патогенез серповидно-клетчной анемии Г У А У→ А Г А А Аминокислота валин глутамин
- 39. Патогенез развития наследственных болезней обмена Отсутствие выработки первичного продукта. Это наиболее частый вариант. В данном случае
- 40. Больной с фенилкетонурией, слабая пигментация кожи, волос, радужной оболочки глаз, умеренная степень олигофрении (Ивар Фелинг, 1934г.)
- 41. Возникновение наследственной болезни обмена веществ НОРМАЛЬНЫЙ МЕТАБОЛИЗМ Субстрат→Фермент+Кофермент→ →Продукт
- 42. Возникновение наследственной болезни обмена веществ НАСЛЕДСТВЕННАЯ БОЛЕЗНЬ ОБМЕНА ВЕЩЕСТВ Субстрат Фермент+Кофермент Продукт метаболит метаболит
- 43. Основные направления терапии наследственных болезней обмена Способы лечения НБО: Ингибиция образования субстрата Коррекция нарушенного баланса метаболитов
- 44. Метаболические пути тирозина и фенилаланина в организме человека
- 45. Муковисцидоз
- 46. Отдельные НБО, для которых разработана диетическая терапия
- 48. Муковисцидоз Барабанные палочки, часовые стекла
- 49. Хромосомные болезни это клинические синдромы, обусловленные изменением числа или структуры хромосом. Частота хромосомных болезней среди новорожденных
- 50. Хромосомные болезни Группы: 1) вызванные изменением числа хромосом (анеуплоидии–изменение количества хромосом какой-то одной пары: моносомия, трисомия
- 51. Хромосомные болезни, обусловленные изменением структуры хромосом: делеция–потеря, дупликация–удвоение, инверсия–разворот на 1800 (происходят внутри хромосомы), транслокация–обмен участками
- 52. Хромосомные болезни Полные формы Мозаичные формы
- 53. Хромосомные перестройки возникают: В аутосомах (с-м Дауна, Патау и др.) и половых хромосомах - гоносомах (с-м
- 54. Хромосомные болезни Особенности хромосомных болезней: 1) множественность поражения - черепно-лицевые дисморфии, врожденные пороки развития внутренних органов,
- 55. Хромосомные болезни 2) Клинически мозаичные формы протекают легче 3) Аутосомные болезни протекают тяжелее, чем аномалии по
- 56. Синдром Дауна (John Down, 1866 г.)
- 57. Синдром Дауна одна из форм геномной патологии кариотип представлен 47 хромосомами поскольку хромосомы 21-й пары, вместо
- 58. Формы синдрома Дауна транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё
- 59. Внешние признаки: «плоское лицо» — 90 % кожная складка на шее у новорожденных — 81 %
- 60. Внешние признаки: мышечная гипотония — 80 % плоский затылок — 78 % короткие конечности — 70
- 61. Внешние признаки аркообразное («готическое») нёбо — 8 % плоская переносица — 52 % бороздчатый язык —
- 62. Внешние признаки : деформация грудной клетки, килевидная или воронкообразная — 27 % пигментные пятна по краю
- 63. Синдром Дауна обезьянья складка норма
- 64. Внешний вид больного с синдромом Дауна
- 65. Синдром Дауна (мозаичная форма)
- 66. Синдром Дауна (мозаичная форма)
- 67. Синдром Дауна (мозаичная форма)
- 68. Синдром Шерешевского-Тернера
- 70. Синдром кошачьего крика (Cri-Du-Chat Syndrome) (синонимы: болезнь кошачьего крика, синдромЛежена по имени описавшего его в 1963
- 71. Синдром Лежена Кариотип 46 ХХ или ХУ, 5р-. Диагноз подтверждается кариологическим исследованием с применением одного из
- 72. Синдром Лежена общее отставание в развитии, низкая масса при рождении и мышечная гипотония, лунообразное лицо с
- 73. Синдром Лежена врожденные пороки сердца, костно-мышечной системы и внутренних органов, микроцефалия, птоз, низкое расположение и деформация
- 74. Синдром Лежена Частота синдрома примерно 1:45000. Соотношение полов М1:Ж1,3 Клиническая картина синдрома и продолжительность жизни людей
- 76. Геномный импринтинг Механизм, с помощью которого различается активность гомологичных генов (или участков хромосом) у индивида в
- 77. Синдром Прадера-Вилли Синдром Энгельмана
- 78. Предполагаемые «болезни импринтинга» у человека Происхождение Синдром Адамса-Оливера материнское Болезнь Альцгеймера отцовское Синдром Энжельмена материнское Врожденный
- 79. Предполагаемые «болезни импринтинга» у человека Поликистоз почек (два локуса) материнское и отцовское Поликистоз яичников материнское Синдром
- 81. Рекомбинация
- 83. Скачать презентацию

Вопросы
1. Общая характеристика моногенных болезней, классификация
2. Патогенез развития наследственных болезней обмена
3.
Вопросы
1. Общая характеристика моногенных болезней, классификация
2. Патогенез развития наследственных болезней обмена
3.

Моногенные заболевания
Распространенность- 1-2% среди новорожденных детей
Описано более 4000 нозологических
Моногенные заболевания
Распространенность- 1-2% среди новорожденных детей
Описано более 4000 нозологических

Моногенные заболевания
Моногенные болезни - это группа наследственных заболеваний, в основе
Моногенные заболевания
Моногенные болезни - это группа наследственных заболеваний, в основе

Мутации
Мутация - любое изменение последовательности ДНК
Мутационая теория де Фриза (1901-1903):
1. Мутации
Мутации
Мутация - любое изменение последовательности ДНК
Мутационая теория де Фриза (1901-1903):
1. Мутации

Моногенные заболевания
Виды генных мутаций
1. Со сдвигом рамки считывания
2. Мутации, обусловленные заменой
Моногенные заболевания
Виды генных мутаций
1. Со сдвигом рамки считывания
2. Мутации, обусловленные заменой

Моногенные заболевания
Виды генных мутаций
По характеру изменений в молекуле ДНК:
Со сдвигом рамки
Моногенные заболевания
Виды генных мутаций
По характеру изменений в молекуле ДНК:
Со сдвигом рамки

Мутация сдвига рамки считывания
Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ
ЦЦА
Мутация сдвига рамки считывания
Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ
ЦЦА

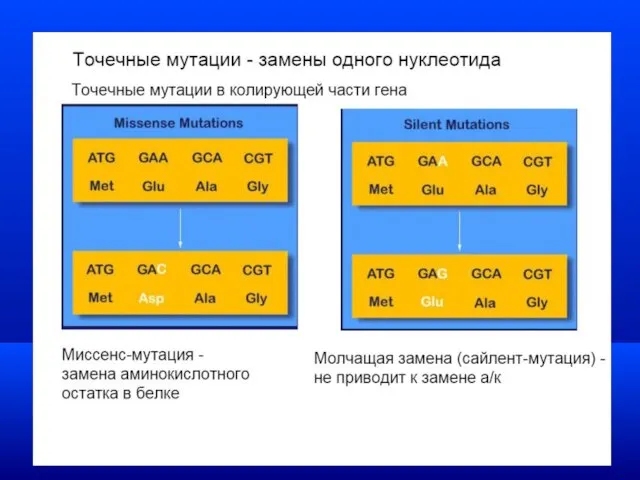
Моногенные заболевания
Виды генных мутаций
Мутации, обусловленные заменой азотистых оснований -
транзиции -замена одного
Моногенные заболевания
Виды генных мутаций
Мутации, обусловленные заменой азотистых оснований -
транзиции -замена одного

Миссенс- мутации
Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ
ЦЦА ЦГГ ТЦГ
Миссенс- мутации
Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ
ЦЦА ЦГГ ТЦГ

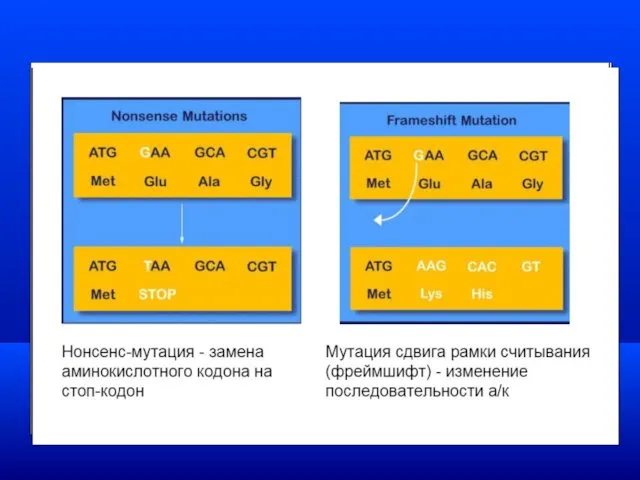
Моногенные заболевания
Если в результате мутаций этой группы образуется
терминирующий кодон (стоп кодон)–УАГ,
Моногенные заболевания
Если в результате мутаций этой группы образуется
терминирующий кодон (стоп кодон)–УАГ,

Нонсенс-мутации
Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ
ЦЦА ЦГГ ТЦГ ЦАГ
Нонсенс-мутации
Нормальная ДНК ГГТ ГЦЦ АГЦ ГТЦ ТАТ
ЦЦА ЦГГ ТЦГ ЦАГ
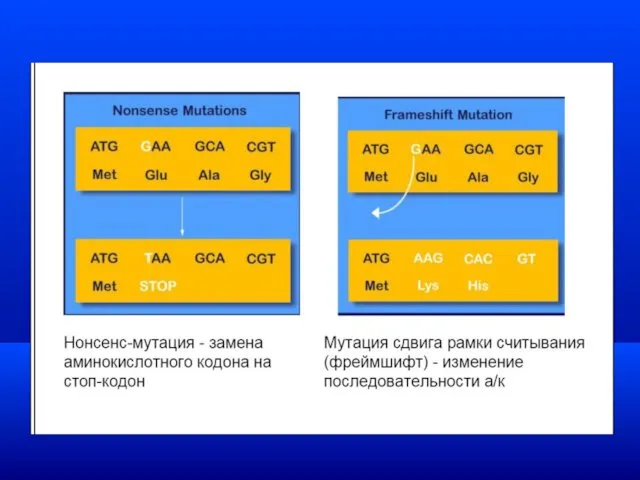

Моногенные заболевания
Генетическая гетерогенность - различия в генетической природе одного заболевания.
Клинический полиморфизм
Моногенные заболевания
Генетическая гетерогенность - различия в генетической природе одного заболевания.
Клинический полиморфизм

Источники генетической гетерогенности :
1) полилокусность (локус – это место расположения гена
Источники генетической гетерогенности :
1) полилокусность (локус – это место расположения гена

Источники генетической гетерогенности
2) полиаллелизм – явление, когда различные мутации одного локуса
Источники генетической гетерогенности
2) полиаллелизм – явление, когда различные мутации одного локуса

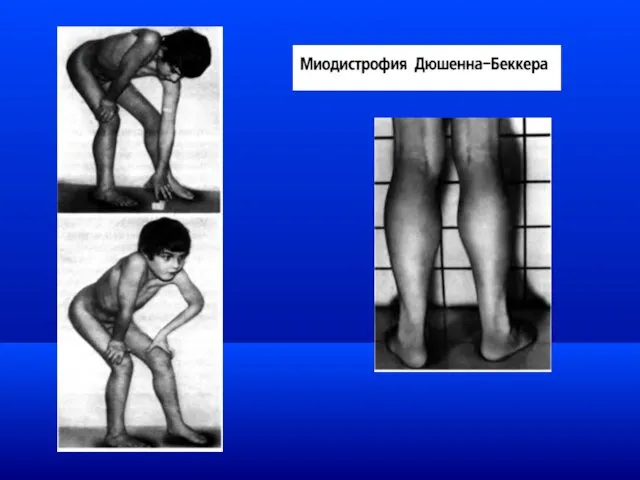
Миодистрофия Дюшенна-Беккера / DMD
Наследственное нервно-мышечное заболевание, характеризующееся прогрессирующими дегенеративными изменениями в
Миодистрофия Дюшенна-Беккера / DMD
Наследственное нервно-мышечное заболевание, характеризующееся прогрессирующими дегенеративными изменениями в

Клинически выделяют две формы миодистрофии:
Миодистрофию Дюшена - мутация в гене (локусе)
Клинически выделяют две формы миодистрофии:
Миодистрофию Дюшена - мутация в гене (локусе)

Фенилкетонурия (ФКУ / PKU)
Врожденное заболевание, вызванное нарушением перехода фенилаланина в тирозин,
Фенилкетонурия (ФКУ / PKU)
Врожденное заболевание, вызванное нарушением перехода фенилаланина в тирозин,
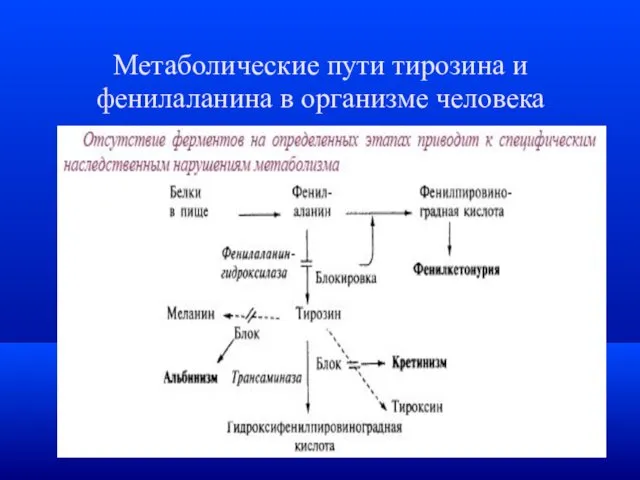
Метаболические пути тирозина и фенилаланина в организме человека
Метаболические пути тирозина и фенилаланина в организме человека

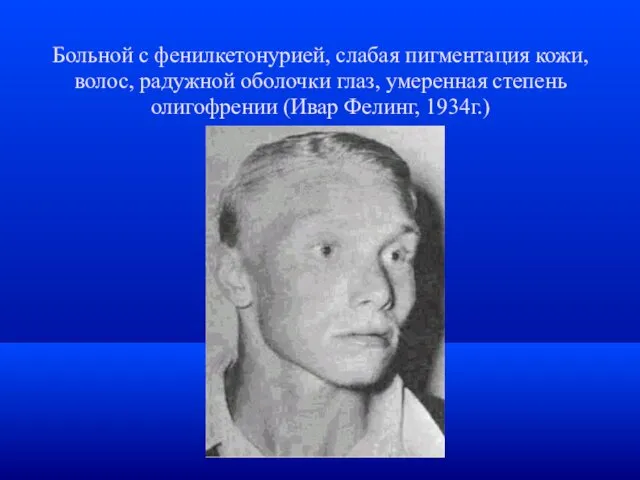
Больной с фенилкетонурией, слабая пигментация кожи, волос, радужной оболочки глаз, умеренная
Больной с фенилкетонурией, слабая пигментация кожи, волос, радужной оболочки глаз, умеренная


Факторы, обусловливающие клинический полиморфизм :
генетическими факторами (полиаллелизм),
модифицирующим влиянием средовых факторов,
сочетанием
Факторы, обусловливающие клинический полиморфизм :
генетическими факторами (полиаллелизм),
модифицирующим влиянием средовых факторов,
сочетанием

Моногенные заболевания
Функции белков
ферментативная,
структурная
транспортная.
Моногенные заболевания
Функции белков
ферментативная,
структурная
транспортная.

Принципы классификации моногенных заболеваний
Генетический
Клинический
Патогенетический
Принципы классификации моногенных заболеваний
Генетический
Клинический
Патогенетический

Моногенные заболевания наследуются по законам Менделя
Болезни с аутосомно-доминантным типом наследования
Болезни с
Моногенные заболевания наследуются по законам Менделя
Болезни с аутосомно-доминантным типом наследования
Болезни с

Классификация моногенных наследственных болезней:
наследственные болезни обмена веществ ( нарушается работа
Классификация моногенных наследственных болезней:
наследственные болезни обмена веществ ( нарушается работа

Клиническая классификация
Заболевания:
Нервные
Нервно-мышечные
Кожные
Глазные
Опорно-двигательного аппарата
Эндокринные
Крови
Сердечно-сосудистой системы
Мочеполовой системы
Желудочно-кишечного тракта
Легких
психические
Клиническая классификация
Заболевания:
Нервные
Нервно-мышечные
Кожные
Глазные
Опорно-двигательного аппарата
Эндокринные
Крови
Сердечно-сосудистой системы
Мочеполовой системы
Желудочно-кишечного тракта
Легких
психические

Наследственные болезни обмена
наследственные дефекты обмена аминокислот (фенилкетонурия, гистидинемия, алкаптонурия, гомоцистинурия).
Наследственные болезни обмена
наследственные дефекты обмена аминокислот (фенилкетонурия, гистидинемия, алкаптонурия, гомоцистинурия).

Наследственные болезни обмена
наследственные дефекты биосинтеза кортикостероидов: адреногенитальный синдром, гипоальдостеронизм и др;
наследственные
Наследственные болезни обмена
наследственные дефекты биосинтеза кортикостероидов: адреногенитальный синдром, гипоальдостеронизм и др;
наследственные

Ферментопатии — наследственные заболевания, обусловленные нарушением синтеза белков ферментов
Ферментопатии — наследственные заболевания, обусловленные нарушением синтеза белков ферментов
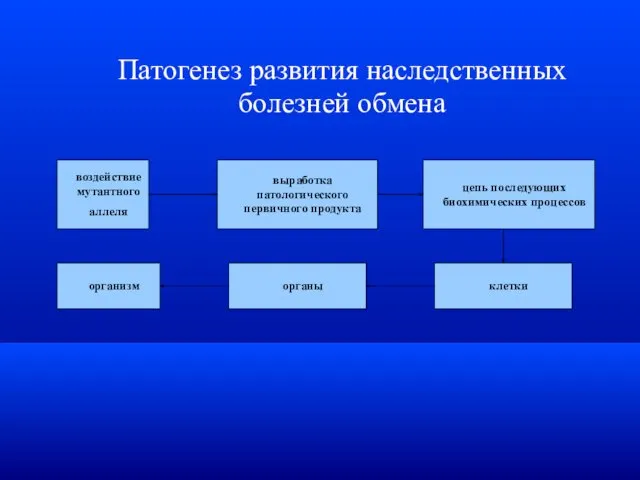
Патогенез развития наследственных болезней обмена
воздействие
мутантного
аллеля
выработка патологического
первичного продукта
цепь последующих
биохимических процессов
клетки
органы
организм
Патогенез развития наследственных болезней обмена
воздействие
мутантного
аллеля
выработка патологического
первичного продукта
цепь последующих
биохимических процессов
клетки
органы
организм

Варианты патологических эффектов мутантных генов
1. Мутантный ген приводит к синтезу избыточного
Варианты патологических эффектов мутантных генов
1. Мутантный ген приводит к синтезу избыточного

Патогенез серповидно-клетчной анемии
Г У А У→ А Г А А
Аминокислота
Патогенез серповидно-клетчной анемии
Г У А У→ А Г А А
Аминокислота

Патогенез развития наследственных болезней обмена
Отсутствие выработки первичного продукта.
Это наиболее частый вариант.
Патогенез развития наследственных болезней обмена
Отсутствие выработки первичного продукта.
Это наиболее частый вариант.
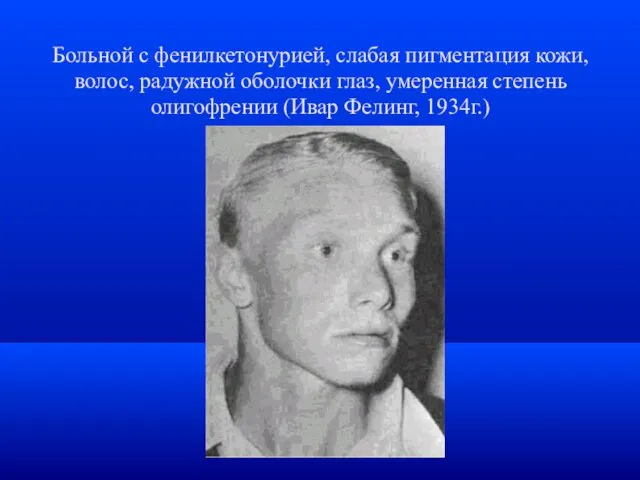
Больной с фенилкетонурией, слабая пигментация кожи, волос, радужной оболочки глаз, умеренная
Больной с фенилкетонурией, слабая пигментация кожи, волос, радужной оболочки глаз, умеренная

Возникновение наследственной болезни обмена веществ
НОРМАЛЬНЫЙ МЕТАБОЛИЗМ
Субстрат→Фермент+Кофермент→
→Продукт
Возникновение наследственной болезни обмена веществ
НОРМАЛЬНЫЙ МЕТАБОЛИЗМ
Субстрат→Фермент+Кофермент→
→Продукт
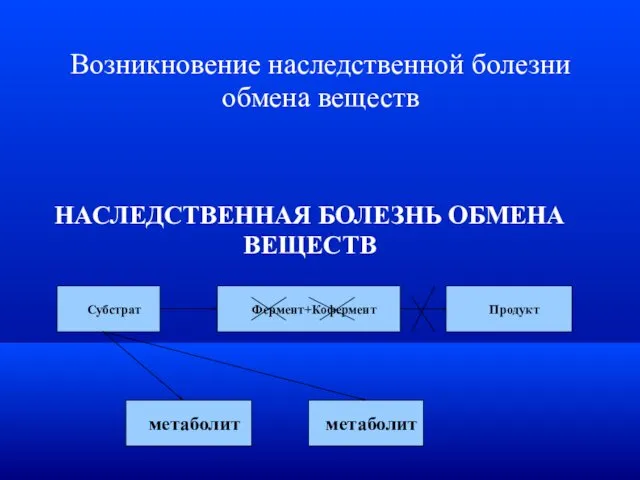
Возникновение наследственной болезни обмена веществ
НАСЛЕДСТВЕННАЯ БОЛЕЗНЬ ОБМЕНА ВЕЩЕСТВ
Субстрат
Фермент+Кофермент
Продукт
метаболит
метаболит
Возникновение наследственной болезни обмена веществ
НАСЛЕДСТВЕННАЯ БОЛЕЗНЬ ОБМЕНА ВЕЩЕСТВ
Субстрат
Фермент+Кофермент
Продукт
метаболит
метаболит

Основные направления терапии наследственных болезней обмена
Способы лечения НБО:
Ингибиция образования субстрата
Коррекция нарушенного
Основные направления терапии наследственных болезней обмена
Способы лечения НБО:
Ингибиция образования субстрата
Коррекция нарушенного

Метаболические пути тирозина и фенилаланина в организме человека
Метаболические пути тирозина и фенилаланина в организме человека
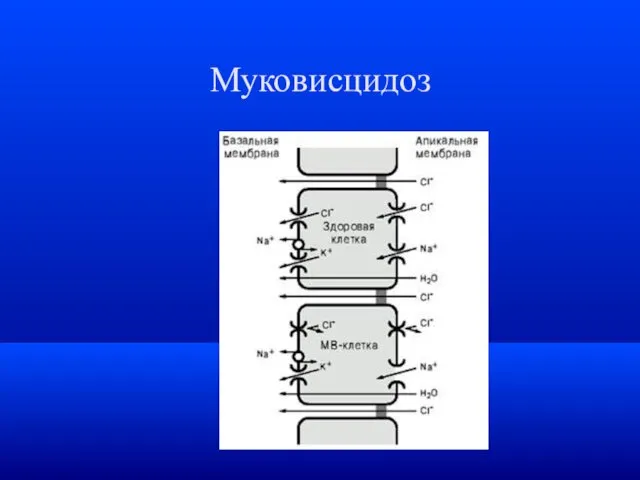
Муковисцидоз
Муковисцидоз
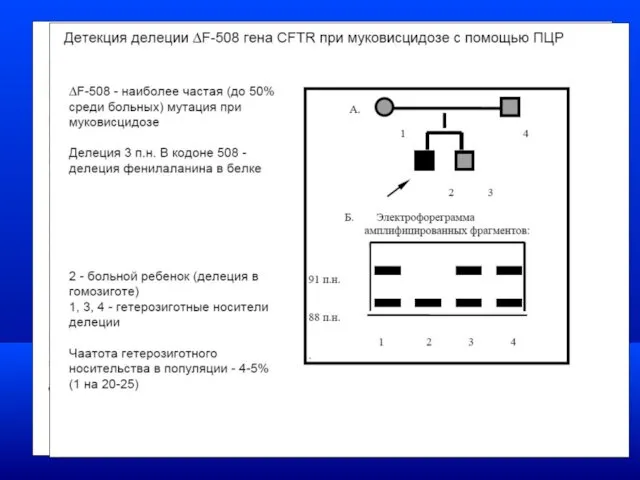
Отдельные НБО, для которых разработана диетическая терапия
Отдельные НБО, для которых разработана диетическая терапия
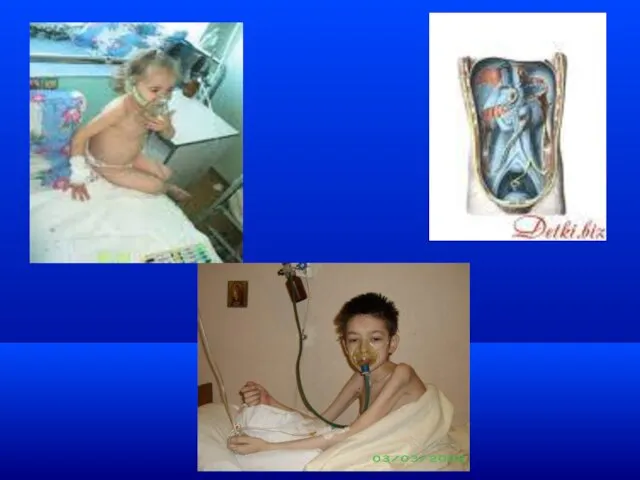
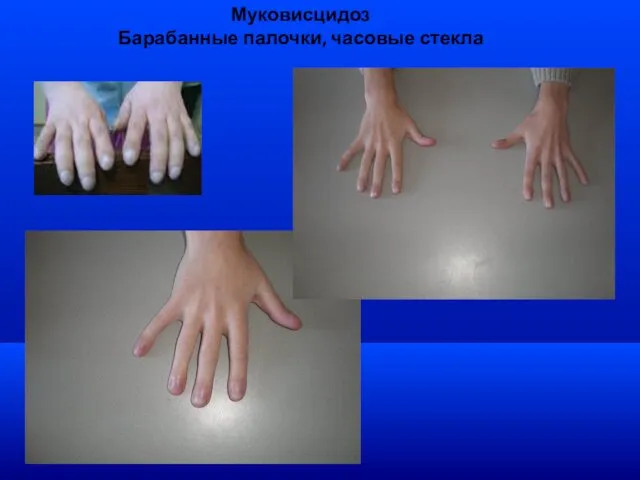
Муковисцидоз
Барабанные палочки, часовые стекла
Муковисцидоз
Барабанные палочки, часовые стекла

Хромосомные болезни
это клинические синдромы, обусловленные изменением числа или структуры хромосом.
Частота хромосомных
Хромосомные болезни
это клинические синдромы, обусловленные изменением числа или структуры хромосом.
Частота хромосомных

Хромосомные болезни
Группы:
1) вызванные изменением числа хромосом (анеуплоидии–изменение количества хромосом какой-то одной
Хромосомные болезни
Группы:
1) вызванные изменением числа хромосом (анеуплоидии–изменение количества хромосом какой-то одной

Хромосомные болезни, обусловленные изменением структуры хромосом:
делеция–потеря,
дупликация–удвоение,
инверсия–разворот на 1800 (происходят внутри хромосомы),
Хромосомные болезни, обусловленные изменением структуры хромосом:
делеция–потеря,
дупликация–удвоение,
инверсия–разворот на 1800 (происходят внутри хромосомы),

Хромосомные болезни
Полные формы
Мозаичные формы
Хромосомные болезни
Полные формы
Мозаичные формы

Хромосомные перестройки возникают:
В аутосомах (с-м Дауна, Патау и др.) и
половых хромосомах
Хромосомные перестройки возникают:
В аутосомах (с-м Дауна, Патау и др.) и
половых хромосомах

Хромосомные болезни
Особенности хромосомных болезней:
1) множественность поражения - черепно-лицевые дисморфии, врожденные пороки
Хромосомные болезни
Особенности хромосомных болезней:
1) множественность поражения - черепно-лицевые дисморфии, врожденные пороки

Хромосомные болезни
2) Клинически мозаичные формы протекают легче
3) Аутосомные болезни протекают тяжелее,
Хромосомные болезни
2) Клинически мозаичные формы протекают легче
3) Аутосомные болезни протекают тяжелее,

Синдром Дауна (John Down, 1866 г.)
Синдром Дауна (John Down, 1866 г.)

Синдром Дауна
одна из форм геномной патологии
кариотип представлен 47 хромосомами поскольку хромосомы 21-й
Синдром Дауна
одна из форм геномной патологии
кариотип представлен 47 хромосомами поскольку хромосомы 21-й

Формы синдрома Дауна
транслокация хромосомы 21 на другие хромосомы (чаще на 15,
Формы синдрома Дауна
транслокация хромосомы 21 на другие хромосомы (чаще на 15,

Внешние признаки:
«плоское лицо» — 90 %
кожная складка на шее у новорожденных — 81 %
монголоидный разрез
Внешние признаки:
«плоское лицо» — 90 %
кожная складка на шее у новорожденных — 81 %
монголоидный разрез

Внешние признаки:
мышечная гипотония — 80 %
плоский затылок — 78 %
короткие конечности — 70 %
открытый рот (в связи
Внешние признаки:
мышечная гипотония — 80 %
плоский затылок — 78 %
короткие конечности — 70 %
открытый рот (в связи

Внешние признаки
аркообразное («готическое») нёбо — 8 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая также
Внешние признаки
аркообразное («готическое») нёбо — 8 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая также

Внешние признаки :
деформация грудной клетки, килевидная или воронкообразная — 27 %
пигментные пятна по
Внешние признаки :
деформация грудной клетки, килевидная или воронкообразная — 27 %
пигментные пятна по
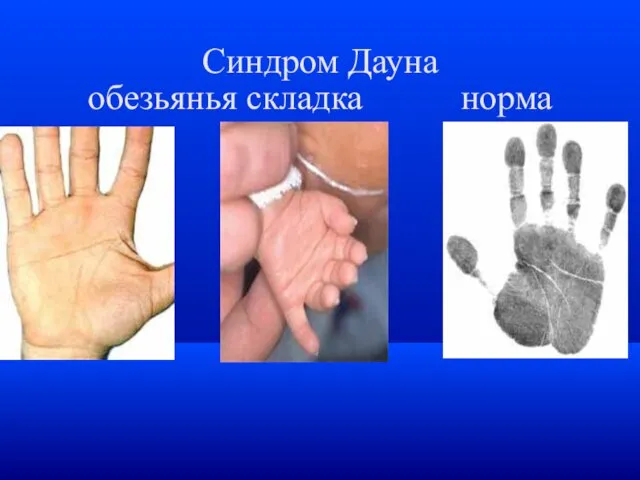
Синдром Дауна
обезьянья складка норма
Синдром Дауна
обезьянья складка норма

Внешний вид больного с синдромом Дауна
Внешний вид больного с синдромом Дауна

Синдром Дауна (мозаичная форма)
Синдром Дауна (мозаичная форма)

Синдром Дауна (мозаичная форма)
Синдром Дауна (мозаичная форма)

Синдром Дауна (мозаичная форма)
Синдром Дауна (мозаичная форма)
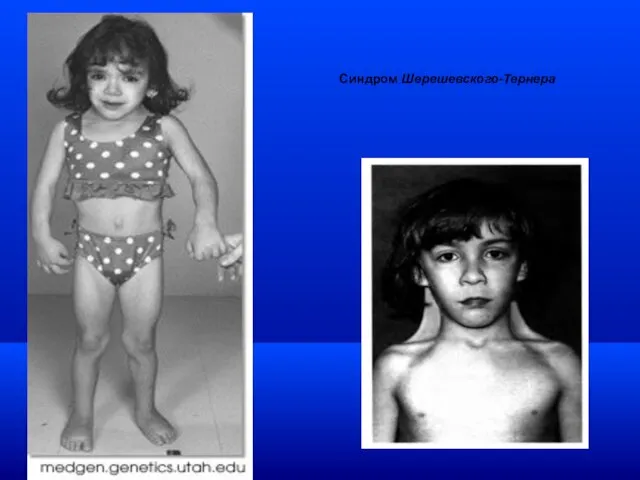
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера
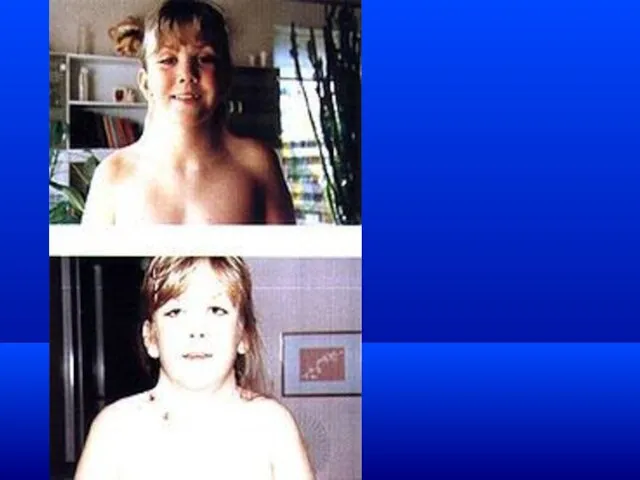

Синдром кошачьего крика (Cri-Du-Chat Syndrome) (синонимы: болезнь кошачьего крика, синдромЛежена по имени описавшего
Синдром кошачьего крика (Cri-Du-Chat Syndrome) (синонимы: болезнь кошачьего крика, синдромЛежена по имени описавшего

Синдром Лежена
Кариотип 46 ХХ или ХУ, 5р-.
Диагноз подтверждается кариологическим исследованием
Синдром Лежена
Кариотип 46 ХХ или ХУ, 5р-.
Диагноз подтверждается кариологическим исследованием

Синдром Лежена
общее отставание в развитии,
низкая масса при рождении и мышечная гипотония,
лунообразное лицо
Синдром Лежена
общее отставание в развитии,
низкая масса при рождении и мышечная гипотония,
лунообразное лицо

Синдром Лежена
врожденные пороки сердца, костно-мышечной системы и внутренних органов,
микроцефалия, птоз,
низкое расположение и
Синдром Лежена
врожденные пороки сердца, костно-мышечной системы и внутренних органов,
микроцефалия, птоз,
низкое расположение и

Синдром Лежена
Частота синдрома примерно 1:45000.
Соотношение полов М1:Ж1,3
Клиническая картина синдрома и продолжительность
Синдром Лежена
Частота синдрома примерно 1:45000.
Соотношение полов М1:Ж1,3
Клиническая картина синдрома и продолжительность
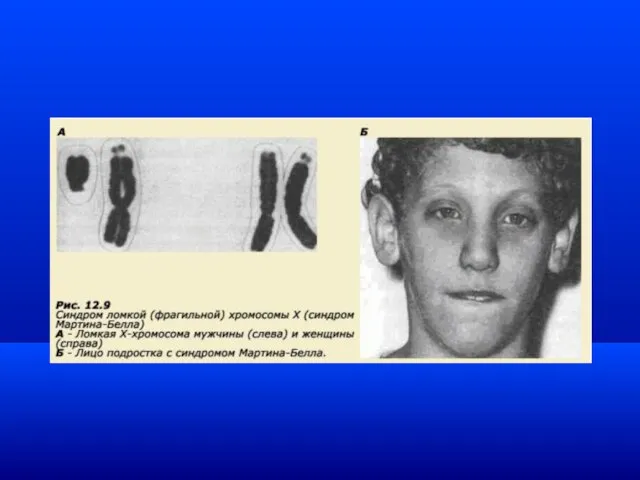

Геномный импринтинг
Механизм, с помощью которого различается активность гомологичных генов (или участков
Геномный импринтинг
Механизм, с помощью которого различается активность гомологичных генов (или участков
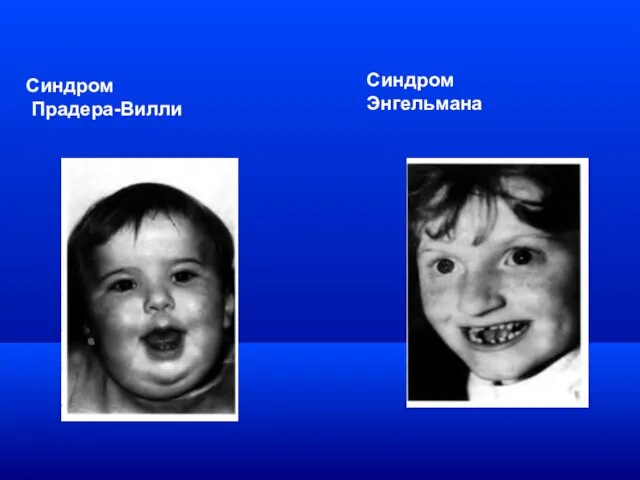
Синдром
Прадера-Вилли
Синдром Энгельмана
Синдром
Прадера-Вилли
Синдром Энгельмана

Предполагаемые «болезни импринтинга» у человека
Происхождение
Синдром Адамса-Оливера материнское
Болезнь Альцгеймера отцовское
Синдром Энжельмена
Предполагаемые «болезни импринтинга» у человека
Происхождение
Синдром Адамса-Оливера материнское
Болезнь Альцгеймера отцовское
Синдром Энжельмена

Предполагаемые «болезни импринтинга» у человека
Поликистоз почек (два локуса) материнское и отцовское
Поликистоз
Предполагаемые «болезни импринтинга» у человека
Поликистоз почек (два локуса) материнское и отцовское
Поликистоз


Рекомбинация
Рекомбинация
 Введение. Биология
Введение. Биология Технология культивирования грибов вешанок и шампиньонов
Технология культивирования грибов вешанок и шампиньонов Генетика
Генетика Особенности Розоцветных Розоцветные (лат. Rosáles) — порядок двудольных растений, состоящий из девяти семейств, самым типичным из
Особенности Розоцветных Розоцветные (лат. Rosáles) — порядок двудольных растений, состоящий из девяти семейств, самым типичным из Тип Круглые черви Урок биологии в 7 классе подготовлен учителем биологии МБОУ СОШ №45 им. А.П. Гайдара города Кирова Куда Елен
Тип Круглые черви Урок биологии в 7 классе подготовлен учителем биологии МБОУ СОШ №45 им. А.П. Гайдара города Кирова Куда Елен Бәсекелестік бір немесе бірнеше түрге
Бәсекелестік бір немесе бірнеше түрге Викторина – игра «Найди лишнее» (группы животных) Автор: Третьякова Елена Васильевна – учитель начальных классов БОУ г.Омска
Викторина – игра «Найди лишнее» (группы животных) Автор: Третьякова Елена Васильевна – учитель начальных классов БОУ г.Омска  Суставы. Соединения костей скелета человека
Суставы. Соединения костей скелета человека Перелетные и зимующие птицы. Конспект занятий в старшей группе
Перелетные и зимующие птицы. Конспект занятий в старшей группе Презентация по биологии Внутреннее строение птиц: пищеварительная, дыхательная, нервная системы
Презентация по биологии Внутреннее строение птиц: пищеварительная, дыхательная, нервная системы  Коммуникация у кошек
Коммуникация у кошек Пути сохранения биологических разнообразий
Пути сохранения биологических разнообразий Разведение лошадей
Разведение лошадей Группа вторичнополостные, целомические животные. Тип кольчатые черви
Группа вторичнополостные, целомические животные. Тип кольчатые черви Витаминдер (Дәрумендер )— денсаулық тірегі
Витаминдер (Дәрумендер )— денсаулық тірегі Эра динозавров в цифрах
Эра динозавров в цифрах Презентация на тему "Звуки леса" - скачать бесплатно презентации по Биологии
Презентация на тему "Звуки леса" - скачать бесплатно презентации по Биологии Молекулярная организация гена
Молекулярная организация гена Презентация на тему "Учение Ч.Дарвина об искусственном отборе" - скачать презентации по Биологии
Презентация на тему "Учение Ч.Дарвина об искусственном отборе" - скачать презентации по Биологии Отдел семенные растения - spermatophyta
Отдел семенные растения - spermatophyta Презентация на тему "Комнатные растения – презентация" - скачать презентации по Биологии
Презентация на тему "Комнатные растения – презентация" - скачать презентации по Биологии Презентация на тему Отдел моховидные
Презентация на тему Отдел моховидные  Презентация на тему "Курение и влияние его на организм" - скачать презентации по Биологии
Презентация на тему "Курение и влияние его на организм" - скачать презентации по Биологии Головоногі молюски
Головоногі молюски  Исследовательская работа Учитель: Томарович Л.А.
Исследовательская работа Учитель: Томарович Л.А. Особенности нуклеотидной последовательности в ДНК эукариот и их функциональное значение
Особенности нуклеотидной последовательности в ДНК эукариот и их функциональное значение Грибы съедобные и ядовитые
Грибы съедобные и ядовитые Презентация кормушки
Презентация кормушки