- Компьютерная эволюционная биология

Содержание
- 2. Понятие координированных замен аминокислотных остатков Исследование наборов гомологичных последовательностей изофункциональных белков является одним из важнейших методов
- 3. Обзор подходов к анализу координированных замен Под координированными заменами аминокислот понимаются за мены двух или нескольких
- 4. Экспериментальные свидетельства кооперативного эффекта замен остатков Тот факт, что стабильность или активность белка зависит от совместного
- 5. В работе [Vemet et al., 1992] проанализированы замены аминокислотных остатков в зоне контакта двух субъединиц папаина.
- 6. Малкольм и сотрудники рассматривали лизоцимы двух видов птиц отряда Курообразных (Galliformes). Анализировались три позиции белка в
- 7. Выявление и анализ координированных замен в последовательностях гомологичных белков. Задача выявления и анализа координированных замен: рассматривается
- 8. Основные методы анализа координированных замен можно условно разделить на две группы. В первую входят методы, которые
- 9. Проблема учета эволюционной зависимости последовательностей. При оценке зависимости аминокислотных замен в парах позиций последовательностей белковых семейств
- 10. Использование информационных мер для оценки парной зависимости аминокислотных замен. При оценке корреляций аминокислотных замен некоторые подходы
- 11. В работе [Clarke, 1995] анализировались последовательности ДНК связывающего домена класса «гомеодомен». Для учета эволюционной зависимости последовательностей
- 12. Метод оценки парных корреляций с использованием информационного подхода был предложен в работе [Korber et al., 1993].
- 13. Этот информационный подход получил дальнейшее развитие в работах Гиро и соавторов, которые использовали в качестве меры
- 14. В целом результаты, полученные О.Б. Птициным и М.В. Волькенштейном, Герштейном и соавторами и Кларк, согласуются с
- 15. АНАЛИЗ РЕЖИМА АДАПТИВНОЙ ЭВОЛЮЦИИ В БЕЛКАХ ВИРУСА ГЕПАТИТА С Вирус гепатита С (ВГС) является основной причиной
- 16. Сравнительный анализ, в частности, позволил выявить шесть основных групп генотипов, обозначаемых цифрами 1-6, а в пределах
- 17. Материалы и методы. Последовательности ВГС. Последовательности для анализа были взяты из базы данных последовательностей белков ВГС
- 18. Выявление адаптивного режима эволюции. Мы использовали критерий отношения скоростей синонимических и несинонимических замен ω= dN/ds, где
- 19. Оценка статистической зависимости между режимом эволюции ко дона и его функциональной нагрузкой. Для оценки статистической зависимости
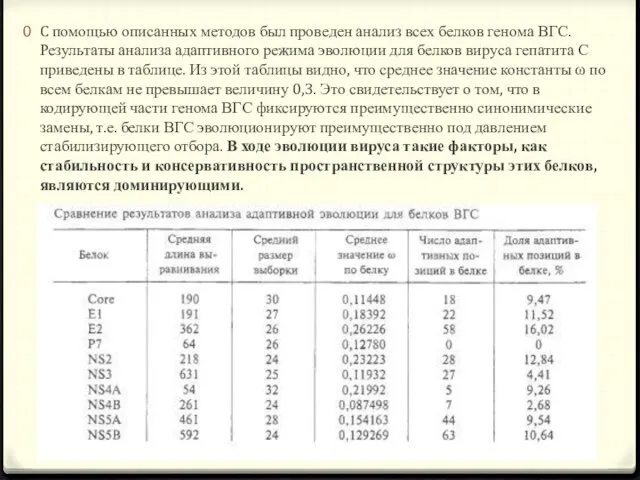
- 20. C помощью описанных методов был проведен анализ всех белков генома ВГС. Результаты анализа адаптивного режима эволюции
- 21. Для всех белков вируса было проведено сопоставление адаптивных позиций с сайтами иммунного ответа и с функциональными
- 22. Из таблицы видно, что большинство белков ВГС содержит позиции, подверженные адаптивной эволюции. Белок Core эволюционирует адаптивно
- 24. Скачать презентацию

Понятие координированных замен аминокислотных остатков
Исследование наборов гомологичных последовательностей изофункциональных белков является
Понятие координированных замен аминокислотных остатков
Исследование наборов гомологичных последовательностей изофункциональных белков является

Обзор подходов к анализу координированных замен
Под координированными заменами аминокислот понимаются за
Обзор подходов к анализу координированных замен
Под координированными заменами аминокислот понимаются за

Экспериментальные свидетельства кооперативного эффекта замен остатков
Тот факт, что стабильность или активность
Экспериментальные свидетельства кооперативного эффекта замен остатков
Тот факт, что стабильность или активность
![В работе [Vemet et al., 1992] проанализированы замены аминокислотных остатков в](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/450513/slide-4.jpg)
В работе [Vemet et al., 1992] проанализированы замены аминокислотных остатков в
В работе [Vemet et al., 1992] проанализированы замены аминокислотных остатков в

Малкольм и сотрудники рассматривали лизоцимы двух видов птиц отряда Курообразных (Galliformes).
Малкольм и сотрудники рассматривали лизоцимы двух видов птиц отряда Курообразных (Galliformes).

Выявление и анализ координированных замен в последовательностях гомологичных белков.
Задача выявления
Выявление и анализ координированных замен в последовательностях гомологичных белков.
Задача выявления

Основные методы анализа координированных замен можно условно разделить на две группы.
Основные методы анализа координированных замен можно условно разделить на две группы.

Проблема учета эволюционной зависимости последовательностей.
При оценке зависимости аминокислотных замен в парах
Проблема учета эволюционной зависимости последовательностей.
При оценке зависимости аминокислотных замен в парах

Использование информационных мер для оценки парной зависимости аминокислотных замен.
При оценке
Использование информационных мер для оценки парной зависимости аминокислотных замен.
При оценке
![В работе [Clarke, 1995] анализировались последовательности ДНК связывающего домена класса «гомеодомен».](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/450513/slide-10.jpg)
В работе [Clarke, 1995] анализировались последовательности ДНК связывающего домена класса «гомеодомен».
В работе [Clarke, 1995] анализировались последовательности ДНК связывающего домена класса «гомеодомен».

Метод оценки парных корреляций с использованием информационного подхода был предложен в
Метод оценки парных корреляций с использованием информационного подхода был предложен в

Этот информационный подход получил дальнейшее развитие в работах Гиро и соавторов,
Этот информационный подход получил дальнейшее развитие в работах Гиро и соавторов,

В целом результаты, полученные О.Б. Птициным и М.В. Волькенштейном, Герштейном и
В целом результаты, полученные О.Б. Птициным и М.В. Волькенштейном, Герштейном и

АНАЛИЗ РЕЖИМА АДАПТИВНОЙ ЭВОЛЮЦИИ В БЕЛКАХ ВИРУСА ГЕПАТИТА С
Вирус гепатита
АНАЛИЗ РЕЖИМА АДАПТИВНОЙ ЭВОЛЮЦИИ В БЕЛКАХ ВИРУСА ГЕПАТИТА С
Вирус гепатита

Сравнительный анализ, в частности, позволил выявить шесть основных групп генотипов, обозначаемых
Сравнительный анализ, в частности, позволил выявить шесть основных групп генотипов, обозначаемых

Материалы и методы.
Последовательности ВГС.
Последовательности для анализа были взяты из базы
Материалы и методы.
Последовательности ВГС.
Последовательности для анализа были взяты из базы

Выявление адаптивного режима эволюции.
Мы использовали критерий отношения скоростей синонимических и несинонимических
Выявление адаптивного режима эволюции.
Мы использовали критерий отношения скоростей синонимических и несинонимических

Оценка статистической зависимости между режимом эволюции ко дона и его функциональной
Оценка статистической зависимости между режимом эволюции ко дона и его функциональной
C помощью описанных методов был проведен анализ всех белков генома ВГС.
C помощью описанных методов был проведен анализ всех белков генома ВГС.
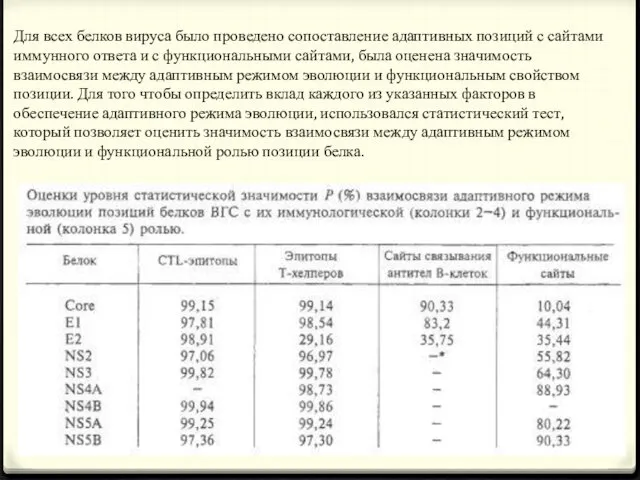
Для всех белков вируса было проведено сопоставление адаптивных позиций с сайтами
Для всех белков вируса было проведено сопоставление адаптивных позиций с сайтами

Из таблицы видно, что большинство белков ВГС содержит позиции, подверженные адаптивной
Из таблицы видно, что большинство белков ВГС содержит позиции, подверженные адаптивной
 Возникновение и эволюция человека
Возникновение и эволюция человека Ядовитые и съедобные грибы
Ядовитые и съедобные грибы Презентация на тему "Антисептика" - скачать бесплатно презентации по Биологии
Презентация на тему "Антисептика" - скачать бесплатно презентации по Биологии Фенологические наблюдения за природой Фенология – наука о сезонном развитии живой природы, обусловленном сменой времен го
Фенологические наблюдения за природой Фенология – наука о сезонном развитии живой природы, обусловленном сменой времен го Презентация на тему "Биотические факторы" - скачать презентации по Биологии
Презентация на тему "Биотические факторы" - скачать презентации по Биологии Пирсинг – красота, требующая жертв? Автор проекта: Шилкин Павел Научный руководитель: учитель высшей квалификационной категории
Пирсинг – красота, требующая жертв? Автор проекта: Шилкин Павел Научный руководитель: учитель высшей квалификационной категории  Внутреннее строение земноводных Учитель биологии МОУ Неклюдовская СОШ Отряскиной Т.А.
Внутреннее строение земноводных Учитель биологии МОУ Неклюдовская СОШ Отряскиной Т.А. Размножение организмов 6 класс - Презентация
Размножение организмов 6 класс - Презентация ОТРУЄННЯ УКУСАМИ ПАВУКІВ.ПЕРША ДОПОМОГА ПРИ УКУСАХ
ОТРУЄННЯ УКУСАМИ ПАВУКІВ.ПЕРША ДОПОМОГА ПРИ УКУСАХ  Дрейф генов в человеческих популяциях
Дрейф генов в человеческих популяциях Биология хомяков, цокоров
Биология хомяков, цокоров Презентация по биологии Физиология почки
Презентация по биологии Физиология почки  Роль биосферы в природе Презентация по биологии
Роль биосферы в природе Презентация по биологии  Охрана животных
Охрана животных Антивірусні засоби Підготувала: Цибульська Вікторія
Антивірусні засоби Підготувала: Цибульська Вікторія  Брожения. Типы жизни, основанные на субстратном фосфорилировании А. Общая характеристика процессов брожения Брожение – это проц
Брожения. Типы жизни, основанные на субстратном фосфорилировании А. Общая характеристика процессов брожения Брожение – это проц Царство бактерии
Царство бактерии Презентация по биологии Отряд Непарнокопытные
Презентация по биологии Отряд Непарнокопытные  Генетическая. Насоедстенность
Генетическая. Насоедстенность Презентация на тему "Ендемічні захворювання" - скачать бесплатно презентации по Биологии
Презентация на тему "Ендемічні захворювання" - скачать бесплатно презентации по Биологии Ауыл шаруашылық дақылдарының өнімділігін жақсартуда қолданылатын микробтық препараттардың ерекшелігі
Ауыл шаруашылық дақылдарының өнімділігін жақсартуда қолданылатын микробтық препараттардың ерекшелігі Оплодотворение
Оплодотворение Где зимуют птицы?
Где зимуют птицы?  Тіндердің шығу тегі мен дамуының заңдылықтары. Тіндердің жіктелуі. Жүйе түзетін факторлар және оның тіндердің тұрақтылығын
Тіндердің шығу тегі мен дамуының заңдылықтары. Тіндердің жіктелуі. Жүйе түзетін факторлар және оның тіндердің тұрақтылығын Охрана природы. Парки и заповедники
Охрана природы. Парки и заповедники Тип Кишечнополостные
Тип Кишечнополостные Focus 4. Human nature. Grammar
Focus 4. Human nature. Grammar Рослинництво світу. Прянощі
Рослинництво світу. Прянощі