- Методы изучения регуляторных районов генов

Содержание
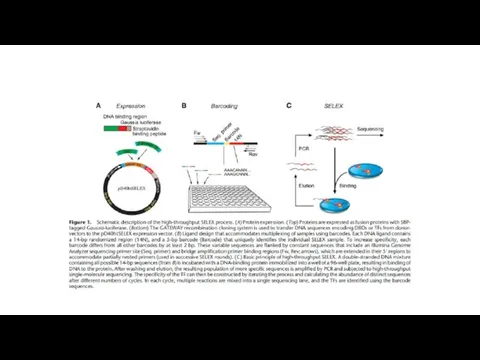
- 2. (N12) 70 mer олигонуклиотид 70 вторая синтезированная цепь Мечение двойной цепи Набор меченых 70bp олигонуклеотидов пропускается
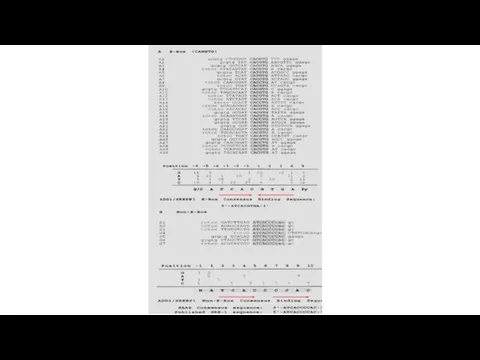
- 8. A МТдт 5'-AGCACTATAGGGACATGATGTTCCACACGTCACATGGTCGTCC-3' МТ27 5'-GATGTCGCGGGAACACAGTGTTCCGTGTACTGTGCAACTACTT-3' 25 5'-GATCCCCCCGGGCATCACCGTGCAGGGGGGGAGGTACAGAGTGTTCT GCGAGGATGCG-3' G/C богатое окружение (28% А/Т п.н.) 111 5'-GATCCTGTTACCATAGTGTAACTTCCTATTCAGGTACAATATGTTCT
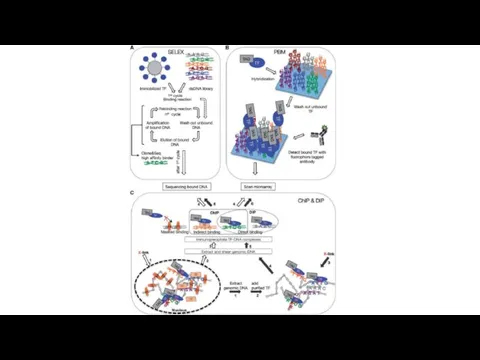
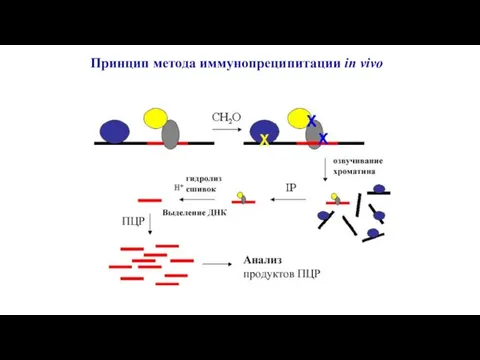
- 9. Принцип метода иммунопреципитации in vivo
- 10. Иммунопреципитация хроматина in vivo с использованием антител против SF-1 для трёх генов мыши Семенники мышей линии
- 11. Scheme of chromatin immunoprecipitation workflow ChIP-chip ChIP-seq microarray analysis Massive parallel sequencing 1 2 3
- 12. Features of p65-binding sites on chromosome 22 R. Martone, G. Euskirchen, P. Bertone, S. Hartman et
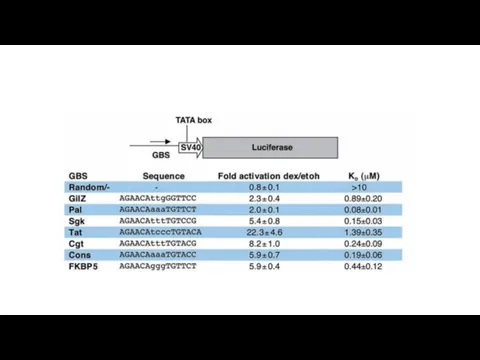
- 13. Location and Position of GREs
- 14. Weak points of TFBS identification by ChIP-seq technology Insufficient resolution Typical ChIP-seq peak – 100-500 bp
- 15. Методы предсказания ССТФ De novo методы поиска ССТФ Методы, основанные на использовании обучающих выборок ССТФ методы
- 16. FoxA2 ChIP-seq в печени мыши [Wederell et al., 2008] Число пиков– 11 475 Средняя высота пика–
- 17. Для создания обучающей выборки из 81 известных ССТФ FoxA использовали данные из базы TRRD и литературных
- 18. Для обучения CHiPMunk были использованы 4455 локуса связывания FoxA2 (CHiP-seq; высота пика >15). Длины мотива для
- 20. Экспериментальная верификация 64 предсказанных ССТФ FoxA методом задержки в геле (EMSA), конкурентный анализ GST-DBD-FoxA2: [32P]-TTR: немеченый
- 21. скор EMSA подтвержденные сайты Скор EMSA расчитывался как отношение угла наклона линейной регрессии log-кривой зависимости от
- 24. Figure 7. Context patterns of the SF-1 BSs recognized by sequence analysis of the top-scoring ChIP-seq
- 27. СУПЕРЭНХАНСЕРЫ
- 28. СУПЕРЭНХАНСЕРЫ
- 29. СУПЕРЭНХАНСЕРЫ
- 31. Скачать презентацию

(N12)
70 mer олигонуклиотид
70 вторая синтезированная цепь
Мечение двойной цепи
Набор меченых 70bp
(N12)
70 mer олигонуклиотид
70 вторая синтезированная цепь
Мечение двойной цепи
Набор меченых 70bp



A
МТдт 5'-AGCACTATAGGGACATGATGTTCCACACGTCACATGGTCGTCC-3'
МТ27 5'-GATGTCGCGGGAACACAGTGTTCCGTGTACTGTGCAACTACTT-3'
25 5'-GATCCCCCCGGGCATCACCGTGCAGGGGGGGAGGTACAGAGTGTTCT
GCGAGGATGCG-3' G/C богатое окружение (28% А/Т п.н.)
A
МТдт 5'-AGCACTATAGGGACATGATGTTCCACACGTCACATGGTCGTCC-3'
МТ27 5'-GATGTCGCGGGAACACAGTGTTCCGTGTACTGTGCAACTACTT-3'
25 5'-GATCCCCCCGGGCATCACCGTGCAGGGGGGGAGGTACAGAGTGTTCT
GCGAGGATGCG-3' G/C богатое окружение (28% А/Т п.н.)
Принцип метода иммунопреципитации in vivo
Принцип метода иммунопреципитации in vivo

Иммунопреципитация хроматина in vivo с использованием
антител против SF-1 для трёх генов
Иммунопреципитация хроматина in vivo с использованием
антител против SF-1 для трёх генов
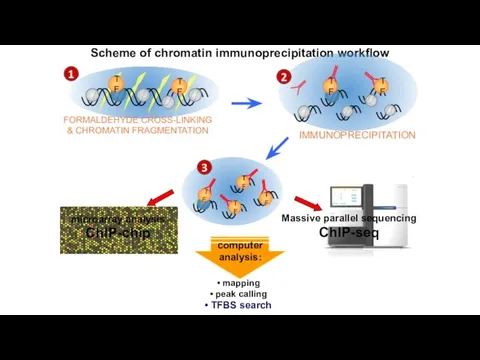
Scheme of chromatin immunoprecipitation workflow
ChIP-chip
ChIP-seq
microarray analysis
Massive parallel sequencing
1
2
3
Scheme of chromatin immunoprecipitation workflow
ChIP-chip
ChIP-seq
microarray analysis
Massive parallel sequencing
1
2
3

Features of p65-binding sites on chromosome 22
R. Martone, G. Euskirchen, P.
Features of p65-binding sites on chromosome 22
R. Martone, G. Euskirchen, P.
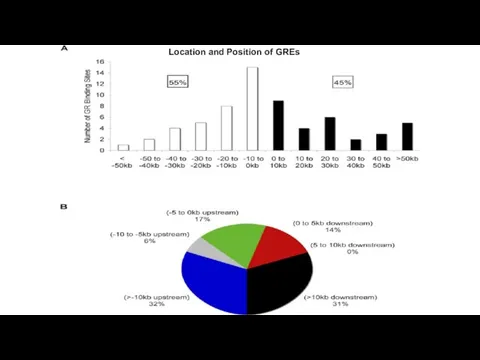
Location and Position of GREs
Location and Position of GREs
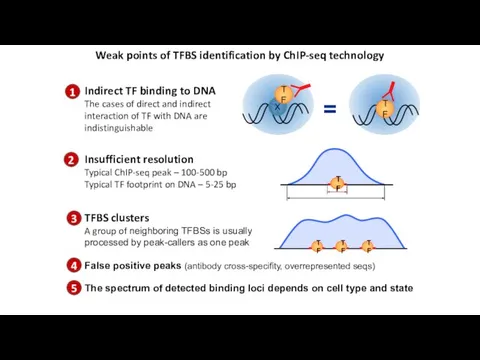
Weak points of TFBS identification by ChIP-seq technology
Insufficient resolution
Typical ChIP-seq peak
Weak points of TFBS identification by ChIP-seq technology
Insufficient resolution
Typical ChIP-seq peak

Методы предсказания ССТФ
De novo методы поиска ССТФ
Методы, основанные на использовании обучающих
Методы предсказания ССТФ
De novo методы поиска ССТФ
Методы, основанные на использовании обучающих
![FoxA2 ChIP-seq в печени мыши [Wederell et al., 2008] Число пиков–](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/506265/slide-15.jpg)
FoxA2 ChIP-seq в печени мыши [Wederell et al., 2008]
Число пиков– 11
FoxA2 ChIP-seq в печени мыши [Wederell et al., 2008]
Число пиков– 11

Для создания обучающей выборки из 81 известных ССТФ FoxA использовали данные
Для создания обучающей выборки из 81 известных ССТФ FoxA использовали данные

Для обучения CHiPMunk были использованы 4455 локуса связывания FoxA2 (CHiP-seq; высота
Для обучения CHiPMunk были использованы 4455 локуса связывания FoxA2 (CHiP-seq; высота

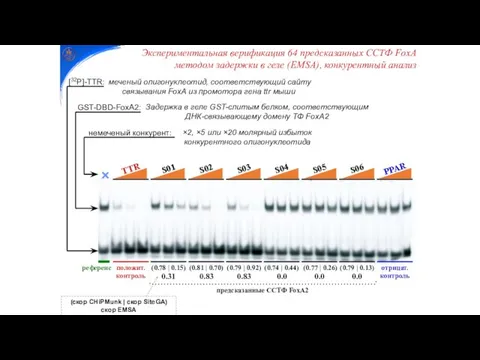
Экспериментальная верификация 64 предсказанных ССТФ FoxA методом задержки в геле (EMSA),
Экспериментальная верификация 64 предсказанных ССТФ FoxA методом задержки в геле (EMSA),
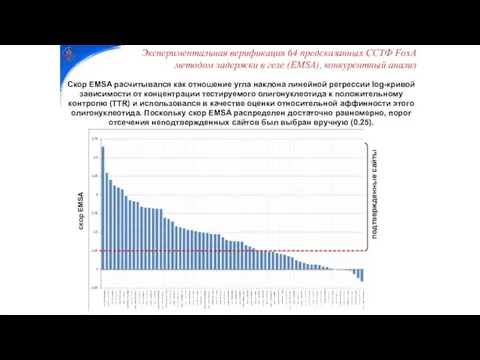
скор EMSA
подтвержденные сайты
Скор EMSA расчитывался как отношение угла наклона линейной регрессии
скор EMSA
подтвержденные сайты
Скор EMSA расчитывался как отношение угла наклона линейной регрессии
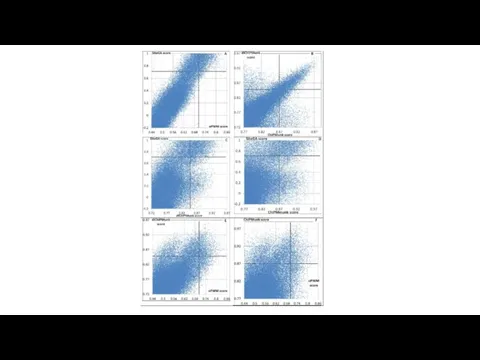
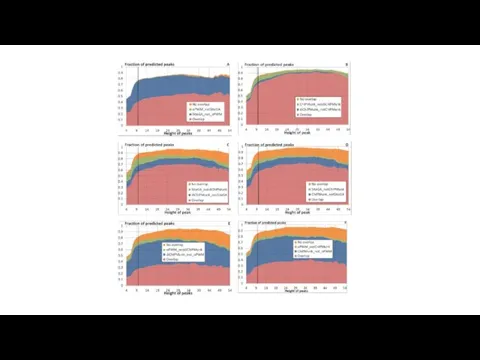

Figure 7. Context patterns of the SF-1 BSs recognized by sequence
Figure 7. Context patterns of the SF-1 BSs recognized by sequence

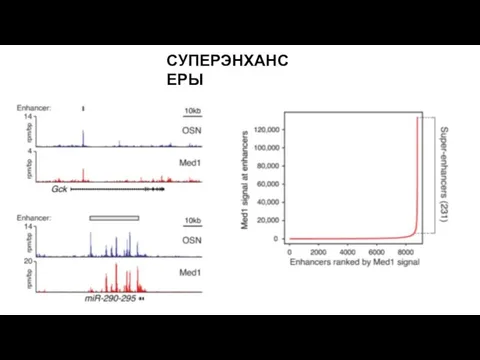
СУПЕРЭНХАНСЕРЫ
СУПЕРЭНХАНСЕРЫ
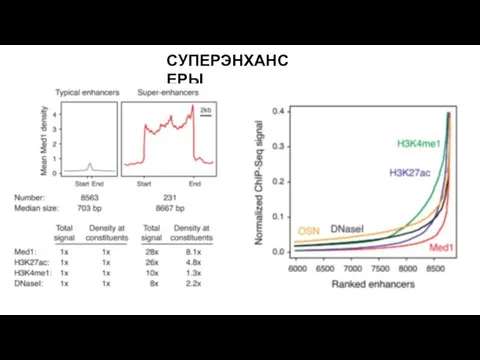
СУПЕРЭНХАНСЕРЫ
СУПЕРЭНХАНСЕРЫ
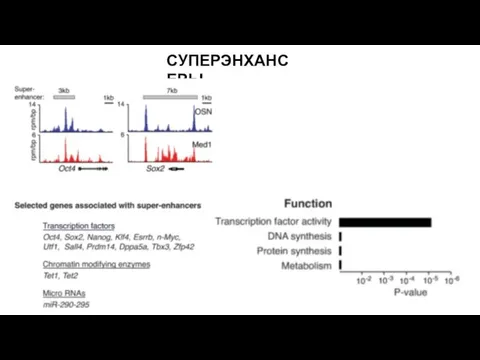
СУПЕРЭНХАНСЕРЫ
СУПЕРЭНХАНСЕРЫ
 Рыхление почвы в цветнике. Подготовка почвы
Рыхление почвы в цветнике. Подготовка почвы Мохообразные
Мохообразные Презентация на тему "Липиды" - скачать презентации по Биологии
Презентация на тему "Липиды" - скачать презентации по Биологии Презентация на тему "Развитие ребенка после рождения" - скачать презентации по Биологии
Презентация на тему "Развитие ребенка после рождения" - скачать презентации по Биологии Морфофункциональные и возрастные особенности опорно - двигательного аппарата
Морфофункциональные и возрастные особенности опорно - двигательного аппарата Химический состав клетки. Рибонуклеиновые кислоты, АТФ
Химический состав клетки. Рибонуклеиновые кислоты, АТФ Кайнозойская эра
Кайнозойская эра Доказательства происхождения человека от животных
Доказательства происхождения человека от животных Анатомо-физиологические особенности нервной системы. Нервный механизм физиологической регуляции функций организма
Анатомо-физиологические особенности нервной системы. Нервный механизм физиологической регуляции функций организма Живое прошлое Земли
Живое прошлое Земли Видообразование. Биология, 11 класс
Видообразование. Биология, 11 класс Системы органов животных
Системы органов животных Шаблоны Ментальных карт
Шаблоны Ментальных карт Урок биологии в 9 классе
Урок биологии в 9 классе Адаптерная теория трансляции
Адаптерная теория трансляции Витамины - это клад
Витамины - это клад Строение и работа сердца и кровеносных сосудов
Строение и работа сердца и кровеносных сосудов Опорно-двигательная аппарат
Опорно-двигательная аппарат Проект: Цветочный путь (благоустройство и озеленение зоны парадного входа школы)
Проект: Цветочный путь (благоустройство и озеленение зоны парадного входа школы) Тема: Історичний розвиток і різноманітність органічного світу Єра динозаврів Виконала: Горовенко Надія
Тема: Історичний розвиток і різноманітність органічного світу Єра динозаврів Виконала: Горовенко Надія  Тема урока: «Цветок, его строение и значение»
Тема урока: «Цветок, его строение и значение» Виды вымирающих животных
Виды вымирающих животных Жизнедеятельность клетки. 6 класс. Урок №7
Жизнедеятельность клетки. 6 класс. Урок №7 Грип. Його природа та можливі ускладнення. Виконала учениця 9-Б класу УФМЛ КНУ ім. Т.Шевченка
Грип. Його природа та можливі ускладнення. Виконала учениця 9-Б класу УФМЛ КНУ ім. Т.Шевченка  Высшая нервная деятельность Поведение
Высшая нервная деятельность Поведение  ТЕМА: Введение ЦЕЛЬ: -Познакомиться с основными целями и задачами физиологии и анатомии человека; -Познакомиться с учеными изу
ТЕМА: Введение ЦЕЛЬ: -Познакомиться с основными целями и задачами физиологии и анатомии человека; -Познакомиться с учеными изу Родина котячі
Родина котячі Анкудинова Юлия 7 «а» класс
Анкудинова Юлия 7 «а» класс