- Методы секвенирования ДНК

Содержание
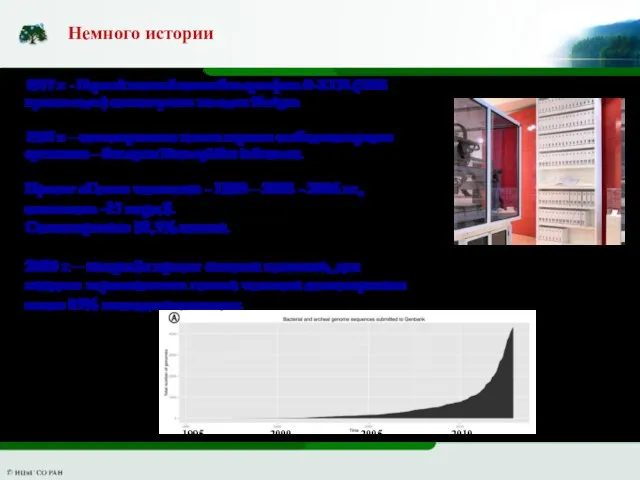
- 2. Немного истории 1977 г. - Первый полный геном бактериофага Φ-X174 (5386 нуклеотидов) секвенирован методом Shotgun 1995
- 3. «Расшифровка» генома Розеттский камень Характерные размеры геномов вирус папиллом человека - 8 т.п.о Escherichia coli -
- 4. Секвенирование по Максаму-Гилберту Химическая деградация ДНК
- 5. Реакция Сэнгера – не ПЦР, а многократный синтез с одной матрицы Секвенирование методом Сэнгера
- 6. 96-capillary ABI 3730XL Первое поколение секвенаторов - капиллярные Нанофор 05 – российская модель
- 7. Начало проекта по секвенированию эукариотического генома А) Выбор объекта Б) Подробная биологическая характеристика объекта, изучение его
- 8. Стратегия секвенирования генома человека 1) 23 хромосомы человека – 23 подпроекта по каждой 2) Субклонирование больших
- 9. Упрощённая схема процесса секвенирования генома методом Shotgun Ферментативные методы фрагментации неприменимы для протяжённых районов. Необходима комбинация
- 10. Подробнее Используемые векторы – ранее М13 phagemid Вентер – производная pBR322, Говорун - pCR2.1 (Invitrogen) Лейшмания
- 11. 1998г. – старт проекта секвенирования первого индивидуального генома человека. Крейг Вентер, компания «Celera Genomics» Использование метода
- 12. Schook et al., 2005 Схема организации геномного проекта на примере генома свиньи (2.6 Gb) А) Секвенирование
- 13. Phred 10 = 90% точность, Phred 20 = 99%, Phred 30 = 99,9%, Phred quality score
- 14. Примеры ошибок и их соотнесение с Phred Компрессия Проскальзывание полимеразы Химерная последовательность - начало конец
- 15. Основную часть ДНК в эукариотических геномах составляют повторы. Это делает крайне затруднительным правильное ассемблирование одиночных последовательностей
- 16. Зависимость числа и длины контигов от покрытия Разрывы в контигах образуются и по чисто статистическим причинам.
- 17. Нелинейность в выборе оптимальных параметров Shotgun Изменения в степени покрытия (%) для 16-kb района при 3-х
- 18. Этапы биоинформатического анализа Собственно сборка секвенированных последовательностей в геном. Структурная аннотация, включающая : идентификацию различных элементов
- 19. Революция цены в геномных исследованиях 454 GS20 1000 Геномов SOLiD 3.0 Illumina GA2
- 21. Скачать презентацию
Немного истории
1977 г. - Первый полный геном бактериофага Φ-X174 (5386
Немного истории
1977 г. - Первый полный геном бактериофага Φ-X174 (5386

«Расшифровка» генома
Розеттский камень
Характерные размеры геномов
вирус папиллом человека - 8 т.п.о
Escherichia coli
«Расшифровка» генома
Розеттский камень
Характерные размеры геномов
вирус папиллом человека - 8 т.п.о
Escherichia coli
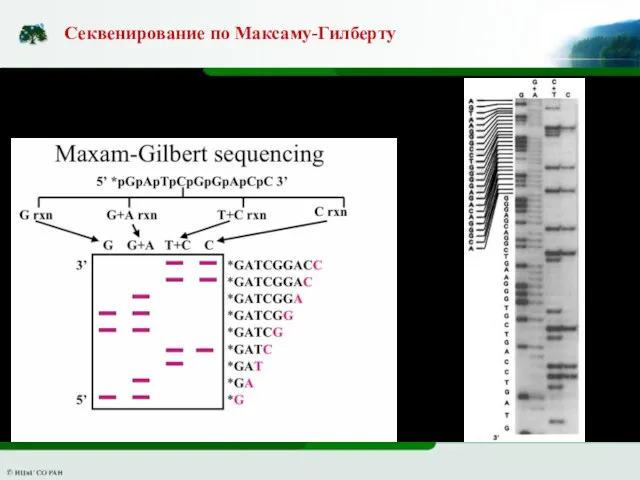
Секвенирование по Максаму-Гилберту
Химическая деградация ДНК
Секвенирование по Максаму-Гилберту
Химическая деградация ДНК
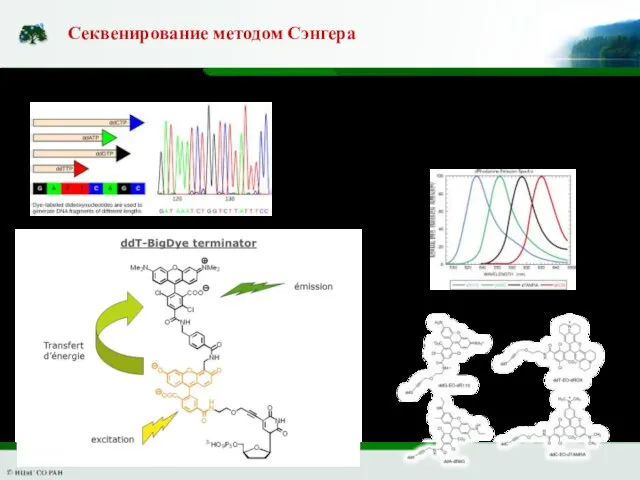
Реакция Сэнгера – не ПЦР, а
многократный синтез с одной матрицы
Реакция Сэнгера – не ПЦР, а
многократный синтез с одной матрицы

96-capillary ABI 3730XL
Первое поколение секвенаторов - капиллярные
Нанофор 05 – российская модель
96-capillary ABI 3730XL
Первое поколение секвенаторов - капиллярные
Нанофор 05 – российская модель
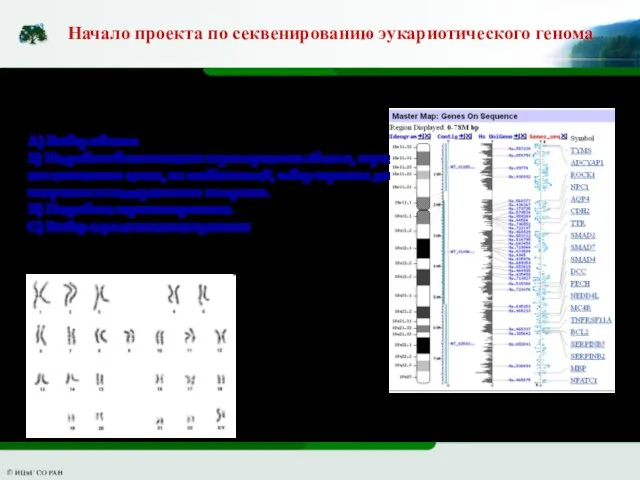
Начало проекта по секвенированию эукариотического генома
А) Выбор объекта
Б) Подробная биологическая характеристика
Начало проекта по секвенированию эукариотического генома
А) Выбор объекта
Б) Подробная биологическая характеристика
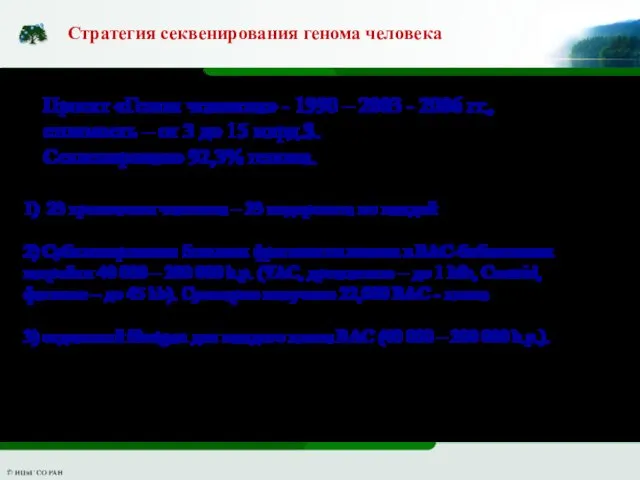
Стратегия секвенирования генома человека
1) 23 хромосомы человека – 23 подпроекта по
Стратегия секвенирования генома человека
1) 23 хромосомы человека – 23 подпроекта по
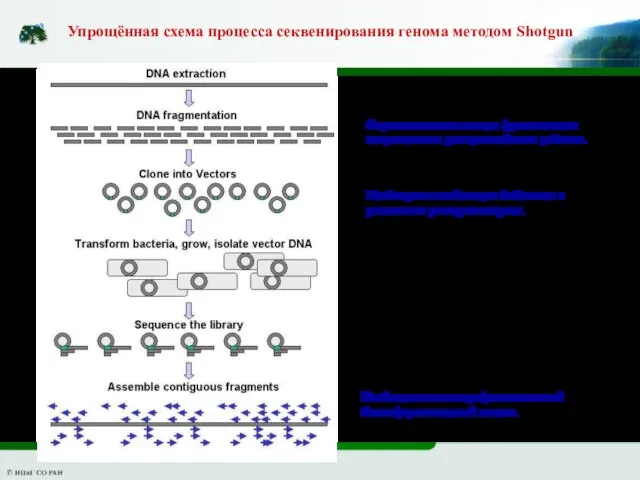
Упрощённая схема процесса секвенирования генома методом Shotgun
Ферментативные методы фрагментации неприменимы
Упрощённая схема процесса секвенирования генома методом Shotgun
Ферментативные методы фрагментации неприменимы

Подробнее
Используемые векторы – ранее М13 phagemid
Вентер – производная pBR322,
Говорун -
Подробнее
Используемые векторы – ранее М13 phagemid
Вентер – производная pBR322,
Говорун -
1998г. – старт проекта секвенирования первого индивидуального генома человека. Крейг Вентер,
1998г. – старт проекта секвенирования первого индивидуального генома человека. Крейг Вентер,
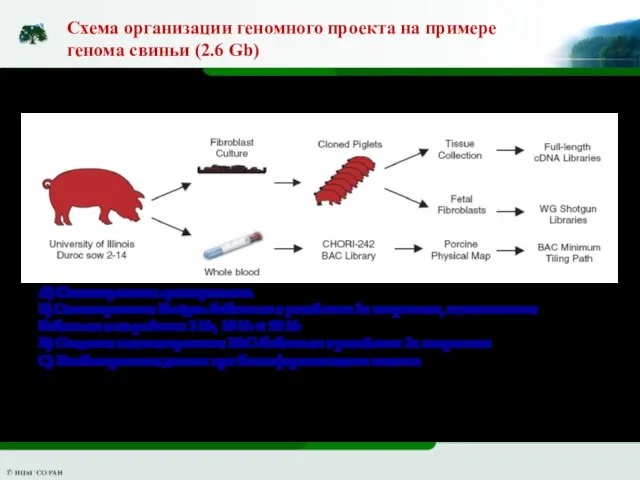
Schook et al., 2005
Схема организации геномного проекта на примере генома свиньи
Schook et al., 2005
Схема организации геномного проекта на примере генома свиньи

Phred 10 = 90% точность, Phred 20 = 99%, Phred 30
Phred 10 = 90% точность, Phred 20 = 99%, Phred 30
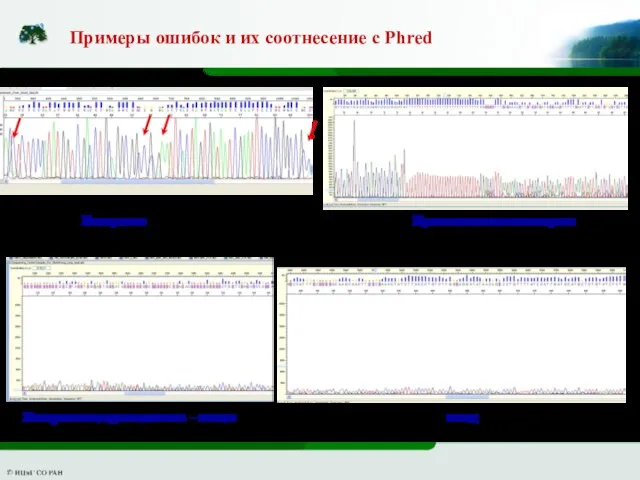
Примеры ошибок и их соотнесение с Phred
Компрессия Проскальзывание полимеразы
Химерная
Примеры ошибок и их соотнесение с Phred
Компрессия Проскальзывание полимеразы
Химерная

Основную часть ДНК в эукариотических геномах составляют повторы. Это делает крайне
Основную часть ДНК в эукариотических геномах составляют повторы. Это делает крайне
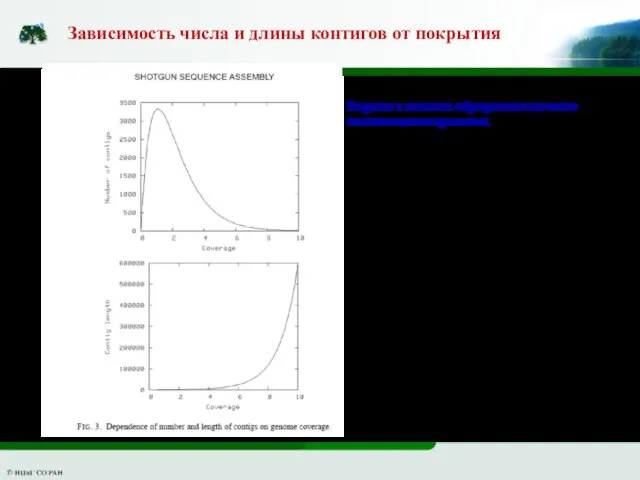
Зависимость числа и длины контигов от покрытия
Разрывы в контигах образуются и
Зависимость числа и длины контигов от покрытия
Разрывы в контигах образуются и
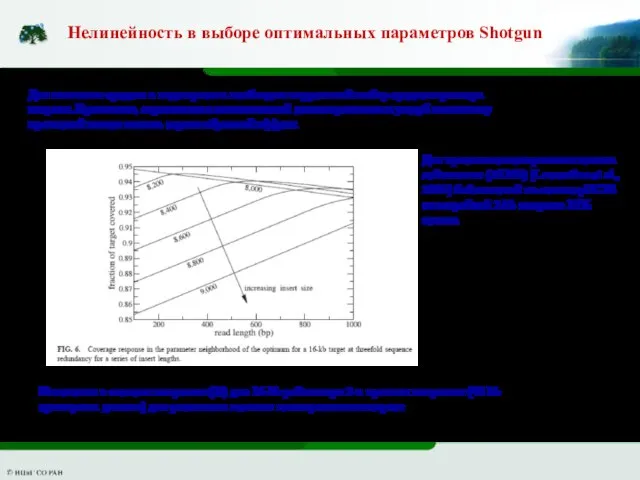
Нелинейность в выборе оптимальных параметров Shotgun
Изменения в степени покрытия (%) для
Нелинейность в выборе оптимальных параметров Shotgun
Изменения в степени покрытия (%) для

Этапы биоинформатического анализа
Собственно сборка секвенированных последовательностей в геном.
Структурная аннотация, включающая :
идентификацию
Этапы биоинформатического анализа
Собственно сборка секвенированных последовательностей в геном.
Структурная аннотация, включающая :
идентификацию
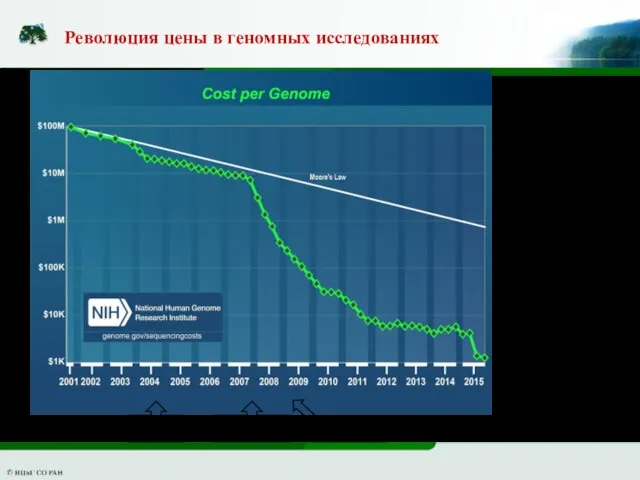
Революция цены в геномных исследованиях
454 GS20
1000 Геномов
SOLiD 3.0
Illumina GA2
Революция цены в геномных исследованиях
454 GS20
1000 Геномов
SOLiD 3.0
Illumina GA2
 Как называются эти пары хромосом?
Как называются эти пары хромосом? Kharari_Sapiens
Kharari_Sapiens Австралопитеки
Австралопитеки Презентация на тему Организм и его свойства
Презентация на тему Организм и его свойства  Жизненный цикл клетки
Жизненный цикл клетки Нервная ткань
Нервная ткань Фотосинтез
Фотосинтез Клетка. Учение о тканях. Виды тканей
Клетка. Учение о тканях. Виды тканей Анатомия, физиология и гигиена человека Анатомия - наука о форме и строении живых организмов
Анатомия, физиология и гигиена человека Анатомия - наука о форме и строении живых организмов Тип Кольчатые черви. Класс Малощетинковые
Тип Кольчатые черви. Класс Малощетинковые Питание растений и грибов
Питание растений и грибов Обобщающее повторение
Обобщающее повторение Антропогенез. Гипотезы возникновения человека. Сходство и различия человека и животных. (Часть 3)
Антропогенез. Гипотезы возникновения человека. Сходство и различия человека и животных. (Часть 3) Процессы торможения в ЦНС
Процессы торможения в ЦНС Функциональная анатомия ствола головного мозга
Функциональная анатомия ствола головного мозга Подцарство простейшие. Амебовые, жгутиконосцы
Подцарство простейшие. Амебовые, жгутиконосцы Бионика и биоинженерия
Бионика и биоинженерия Презентация на тему Биология – наука о живой природе
Презентация на тему Биология – наука о живой природе Роль гормонів у фізичному, психічному і статевому розвитку
Роль гормонів у фізичному, психічному і статевому розвитку Гаметогенез, оплодотворение
Гаметогенез, оплодотворение Презентация по биологии Почвенная среда обитания
Презентация по биологии Почвенная среда обитания  Грибы-паразиты. Головня
Грибы-паразиты. Головня Фотосинтез
Фотосинтез Дослідження росту вегетативних органів рослин
Дослідження росту вегетативних органів рослин Міні – проект на тему: Опале листя. Це користь чи шкода?
Міні – проект на тему: Опале листя. Це користь чи шкода? Овогенез. Сперматогенез
Овогенез. Сперматогенез Витамины
Витамины Презентация на тему "Современное представление о механизмах и закономерностях эволюции" - скачать презентации по Биологии
Презентация на тему "Современное представление о механизмах и закономерностях эволюции" - скачать презентации по Биологии