- Презентация на тему "Наследственные болезни человека" - скачать презентации по Биологии

Содержание
- 2. Наследственные болезни - заболевания человека, обусловленные хромосомными и генными мутациями. Их более 6000 Нередко ошибочно термины
- 3. При возникновении мутации в клетке на ранних стадиях онтогенеза, из неё будут развиваться ткани, все клетки
- 4. Генеративные мутации Моногенные - мутации в одном гене Общая частота генных болезней в популяции составляет 1-2%
- 5. НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ ГЕННЫЕ БОЛЕЗНИ
- 7. КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ Генные болезни; Хромосомные болезни; Болезни с наследственной предрасположенностью (мультифакториальные болезни); Группа генетических болезней,
- 8. ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ Ранняя манифестация; Хроническое прогредиентное течение; Относительная резистентность к терапии; Множественность поражения;
- 9. Генные болезни - это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне. Число
- 10. Нейрофиброматоз Синдром Марфана Болезнь Олбрайта Дизостозы Отосклероз Пароксизмальная миоплегия Талассемия Семейная гипехолестеринемия Несовершенный остеогенез Болезнь Гентингтона
- 11. мышечная дистрофия типа Дюшенна, гемофилии А и В, синдрома Леша — Найхана, болезни Гунтера, болезни Фабри
- 12. СИНДРОМ МАРФАНА Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани, впервые описана в 1886
- 13. Синдром Марфана - наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким ростом с относительно коротким туловищем,
- 14. Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению
- 15. Сидндром Марфана Наследственное заболевание соединительной ткани , проявляющееся изменениями скелета: высоким ростом с относительно коротким туловищем
- 16. СИНДРОМ МАРФАНА Симптом «большого пальца» Арахнодактелия (длинные пальцы) Симптом «запястья» «Сандалевидная щель»
- 18. Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечнососудистых осложнений, но и появлению
- 19. МАРФАНА СИНДРОМ Впервые описан в 1896 г. Клинические признаки: высокий рост, арахнодактилия, подвывих хрусталика, порок митрального
- 20. Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению
- 21. Симптомы «Большого пальца» и «запястья» Арахнодактилия
- 22. Синдром Марфана
- 24. Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от аневризма аорты. Единственная компенсация –
- 25. Известные люди с синдромом Марфана Эхнатон, Паганини Эхнатон, Паганини Эхнатон Н. Паганини Ш. де Голль А.
- 26. ФЕНИЛКЕТОНУРИЯ (ФЕНИЛПИРОВИНОГРАДНАЯ ОЛИГОФРЕНИЯ) Фенилкетонурия (ФКУ) – это она из самых частых форм наследственных дефектов обмена аминокислот.
- 27. Классическая форма фенилкетонурии, ребенок в возрасте 1 год 7 мес. Грубая задержка психомоторного развития, судороги (до
- 29. Несовершенный остеогенез наследуется по аутосомно-рецессивному типу. Несовершенный остеогенез
- 30. Несовершенный остеогенез фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями в суставах, глухотой, голубыми склерами,
- 31. Несовершенный остеогенез
- 32. Ахондроплазия — врожденное поражение скелета — врождённая болезнь, характеризующаяся нарушением развития хрящевой ткани; проявляется карликовостью, короткими
- 33. Человек, закончивших свой рост, достигает 30 - 41 см.
- 34. Причины мутации в настоящее время не известны.
- 35. Знакомьтесь, это Джиоти Амге - 18-летняя студентка из Индии. Еще с детства она перестала расти из-за
- 37. СИНДРОМ ХОЛТ – ОРАМА (СИНДРОМ РУКА – СЕРДЦЕ) Синдром Холт – Орама представляет собой моногенный синдром
- 38. Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую
- 39. МУКОВИСЦИДОЗ (КИСТОФИБРОЗ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ) Заболевание обусловлено генерализованным поражением экзокринных желез. Частота муковисцедоза среди новорожденных в европейской
- 40. Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы и печени, характеризующееся, в первую
- 41. Новорожденный с муковисцидозом
- 42. Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве, хотя в 4% случаев диагноз
- 43. Распространенность заболевания сильно отличается в разных этнических группах. Среди белого населения Северной Америки и Северной Европы
- 44. Гомоцистинурия – нарушение метаболизма метионина. При гомоцистинурии, как и при других наследственных нарушениях обмена веществ, в
- 45. Ира Д., 7 лет. Диагноз: гомоцистинурия
- 46. Подвывих хрусталика при гомоцистинурии
- 47. Материнская гистидинемия. Нарушение обмена фермента гистидина Описана в 1974 году Леоном у четверых детей, родившихся у
- 48. Вадим 3 года родился без особенностей. С введением прикорма появилась общая мышечная гипотония, расстройства глотания, голову
- 49. СИНДРОМ ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ Относится к редким заболеваниям, для которого характерен своеобразный симтомокомплекс – сочетание пигментного ретинита, ожирение,
- 50. Дети с синдромом ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ полидактилия
- 51. Наследственные болезни обмена веществ соединительной ткани. МУКОПОЛИСАХАРИДОЗЫ Под термином «мукополисахаридозы» объединяется ряд патологических процессов, в основе
- 52. МУКОПОЛИСАХАРИДОЗ I ТИПА (СИНДРОМ ГУРЛЕР) Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной умственной отсталостью, помутнением роговицы
- 53. а - синдром Гурлер (большая голова, грубые черты лица, широкая запавшая переносица, увеличенное расстояние между глазами,
- 54. МУКОПОЛИСАХАРИДОЗ II ТИПА (СИНДРОМ ГУНТЕРА) В клинической картине на первый план выступает костные деформации и тугоподвижность
- 55. МУКОПОЛИСАХАРИДОЗ III (А, В) ТИПА (СИНДРОМ САНФИЛИППО) МУКОПОЛИСАХАРИДОЗ IV ТИПА (СИНДРОМ МОРКИО) В клинической картине заболевания
- 56. б — синдром Моркио (килевидная деформация грудной клетки, кифоз поясничного отдела позвоночника, множественные деформации суставов, сгибательные
- 57. Изменения костной ткани при мукополисахаридозах
- 58. МУКОПОЛИСАХАРИДОЗ V ТИПА (СИНДРОМ ШЕЙЕ) МУКОПОЛИСАХАРИДОЗ VI ТИПА (СИНДРОМ МАРОТО-ЛАМИ) Клиническая картина заболевания складывается из наличия
- 59. Больная с синдромом Шейе
- 60. в - синдром Шейе (грубые черты лица, запавшая переносица, короткая шея, множественные деформации суставов, в том
- 61. Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных преждевременным
- 62. ПРОГЕРИЯ Описана в 1886 г. Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения в 8-10 раз.
- 65. Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по типу гиперкератоза, проявляется образованием
- 70. НЕЙРОФИБРОМАТОЗ (БОЛЕЗНЬ РЕКЛИНГХАУЗЕНА) Известно семь нозологических форм нейрофиброматоза (НФ), среди которых НФ – 1 (периферический нейрофиброматоз)
- 71. Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по размеру и окраске, часто кофе
- 72. Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное заболевание, характеризующееся наличием множественных пигментных
- 73. Нейрофиброматоз У больного этим заболеванием наблюдается: слоновость левой верхней конечности, обусловленная множественными нейрофиброматозными узлами.
- 74. Почти у всех больных на радужке обнаруживаются небольшие гамартомы . Нейрофибромы могут подвергаться злокачественной трансформации с
- 75. Мутация, за развитие этого типа, обнаружена в хромосоме 17, в зоне, кодирующей белок нейрофибромин, подавляющий рост
- 76. НЕЙРОФИРОМАТОЗ Множественные нейрофибромы
- 80. АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (ВРАЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ) Адреногенитальный синдром (АГС) относится к группе наследственных нарушений биосинтеза стероидных
- 81. Новорожденная девочка с двойственным строением наружных половых органов Мальчики шести лет (справа – опережение полового и
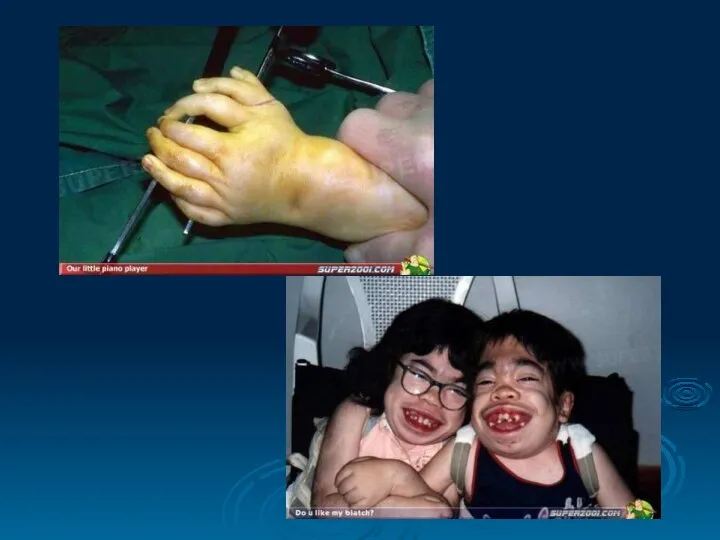
- 84. ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ДЮШЕННА) Это одна из самых частых форм наследственных нервно – мышечных заболеваний. Впервые
- 85. А,Б – Ряд последовательных движений при принятии вертикального положения симптом «лестницы» А Б В В –
- 88. Странное племя людей-страусов (сапади) в Центральной Африке отличает от прочих обитателей Земли удивительное свойство: на ногах
- 90. Синдром Ангельмана Синдром Ангельмана (СА) - это нейро-генетическое заболевание, характеризующееся интеллектуальной и физической задержкой развития, нарушениями
- 94. Дети наркоманов. Копия, воск.
- 95. Сиамские близнецы, у родителей-наркоманов. Натура, заспиртованные.
- 96. Дети у родителей больных наследственными заболеваниями. Копия, воск.
- 97. Ребенок, родившийся в результате инцеста(кровосмешения родственников). Натура, заспиртован. Ответьте на проблемный вопрос. Почему в близкородственных браках
- 98. Ребенок, родившийся в семье чернобыльцев. Натура, мумия.
- 99. Человек-циклоп, и женщина-слон. Жили в 19 веке. Копия, воск.
- 100. Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом, можно говорить о многообразии видов
- 101. Если принять, что у человека примерно 100 000 генов и каждый ген может мутировать и контролировать
- 102. Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации происходят постоянно как отрицательные, так
- 104. Скачать презентацию
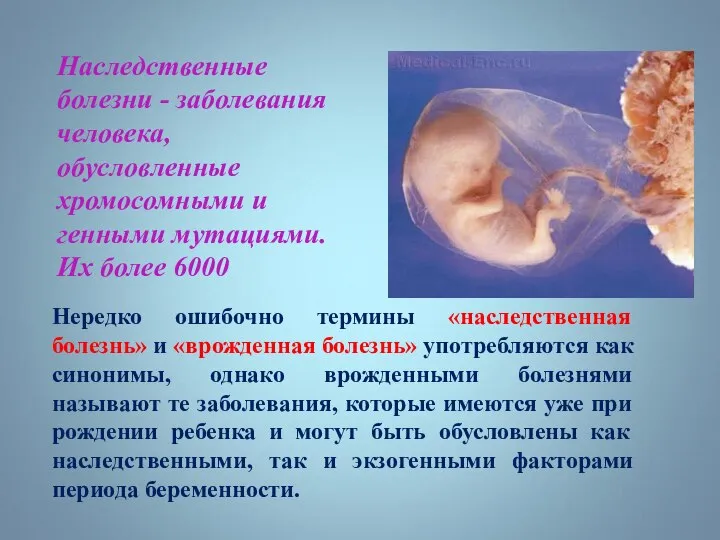
Наследственные болезни - заболевания человека, обусловленные хромосомными и генными мутациями. Их более
Наследственные болезни - заболевания человека, обусловленные хромосомными и генными мутациями. Их более
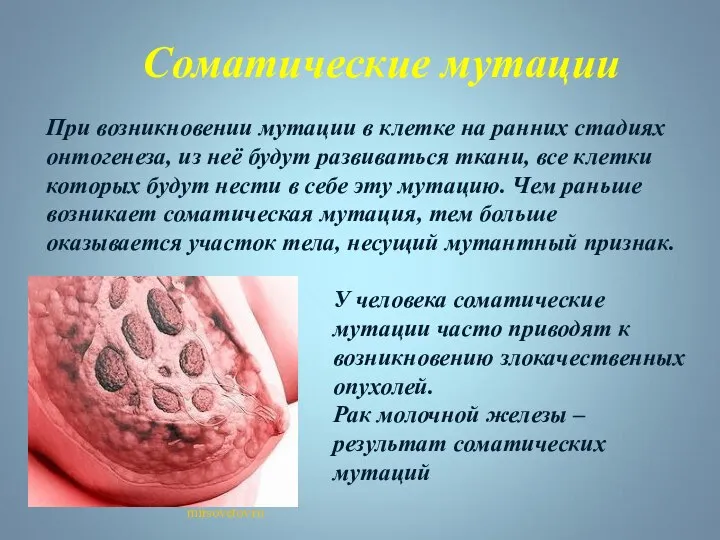
При возникновении мутации в клетке на ранних стадиях онтогенеза, из неё
При возникновении мутации в клетке на ранних стадиях онтогенеза, из неё

Генеративные мутации
Моногенные - мутации в одном гене
Общая частота генных болезней в
Генеративные мутации
Моногенные - мутации в одном гене
Общая частота генных болезней в
НАСЛЕДСТВЕННАЯ
ПАТОЛОГИЯ
ГЕННЫЕ БОЛЕЗНИ
НАСЛЕДСТВЕННАЯ
ПАТОЛОГИЯ
ГЕННЫЕ БОЛЕЗНИ


КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Генные болезни;
Хромосомные болезни;
Болезни с наследственной предрасположенностью (мультифакториальные болезни);
Группа
КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Генные болезни;
Хромосомные болезни;
Болезни с наследственной предрасположенностью (мультифакториальные болезни);
Группа

ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Ранняя манифестация;
Хроническое прогредиентное течение;
Относительная резистентность к терапии;
Множественность
ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Ранняя манифестация;
Хроническое прогредиентное течение;
Относительная резистентность к терапии;
Множественность

Генные болезни - это разнородная по клиническим проявлениям группа заболеваний, обусловленных

Нейрофиброматоз
Синдром Марфана
Болезнь Олбрайта
Дизостозы
Отосклероз
Пароксизмальная миоплегия
Талассемия
Семейная гипехолестеринемия
Несовершенный остеогенез
Болезнь Гентингтона
Поликистоз почек
Нейрофиброматоз
Синдром Марфана
Болезнь Олбрайта
Дизостозы
Отосклероз
Пароксизмальная миоплегия
Талассемия
Семейная гипехолестеринемия
Несовершенный остеогенез
Болезнь Гентингтона
Поликистоз почек

мышечная дистрофия типа Дюшенна, гемофилии А и В, синдрома Леша — Найхана,
мышечная дистрофия типа Дюшенна, гемофилии А и В, синдрома Леша — Найхана,
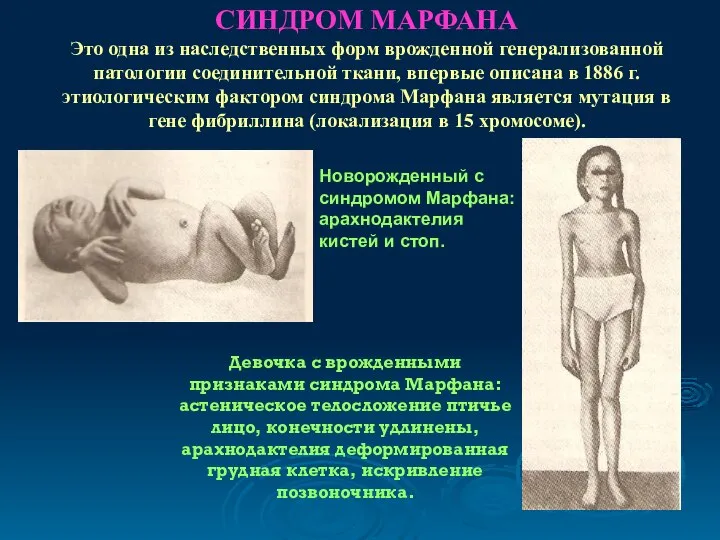
СИНДРОМ МАРФАНА
Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани,
СИНДРОМ МАРФАНА Это одна из наследственных форм врожденной генерализованной патологии соединительной ткани,
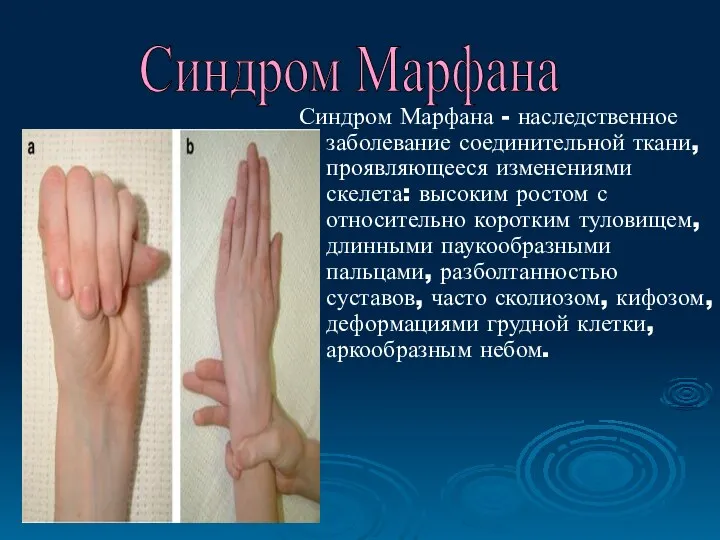
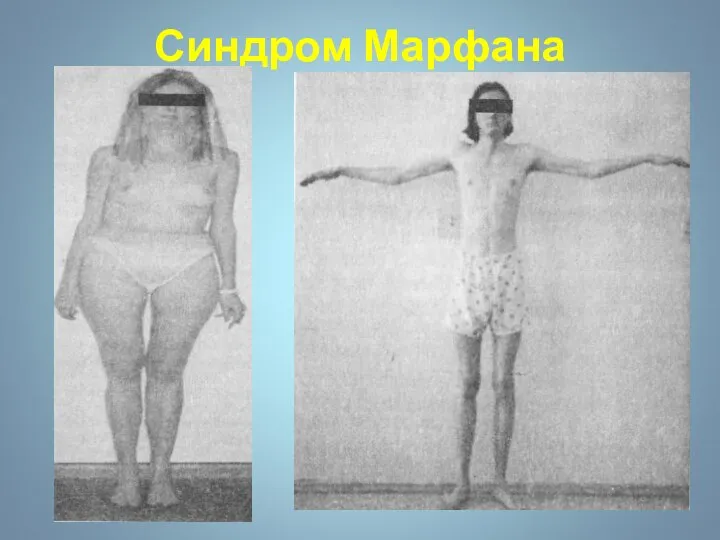
Синдром Марфана - наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким
Синдром Марфана - наследственное заболевание соединительной ткани, проявляющееся изменениями скелета: высоким
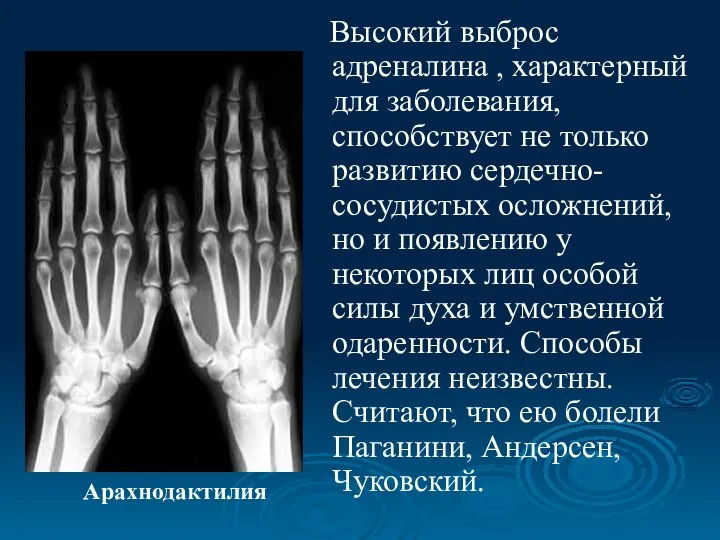
Высокий выброс адреналина , характерный для заболевания, способствует не только
Высокий выброс адреналина , характерный для заболевания, способствует не только

Сидндром Марфана
Наследственное заболевание соединительной ткани , проявляющееся изменениями скелета: высоким
Сидндром Марфана
Наследственное заболевание соединительной ткани , проявляющееся изменениями скелета: высоким
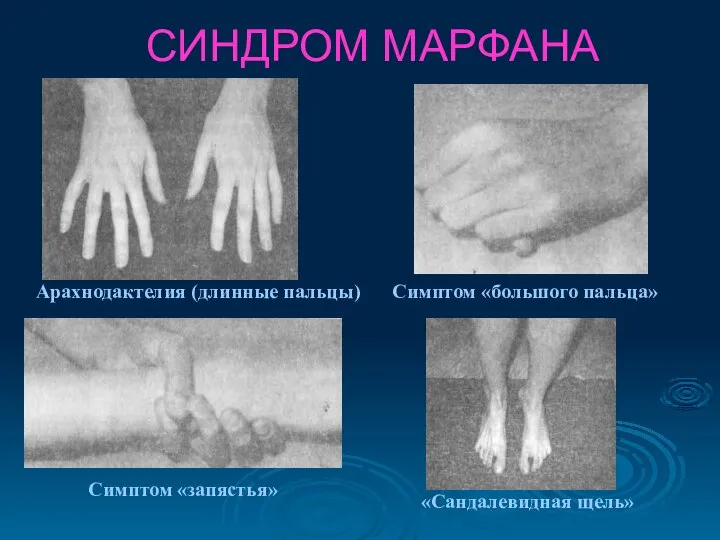
СИНДРОМ МАРФАНА
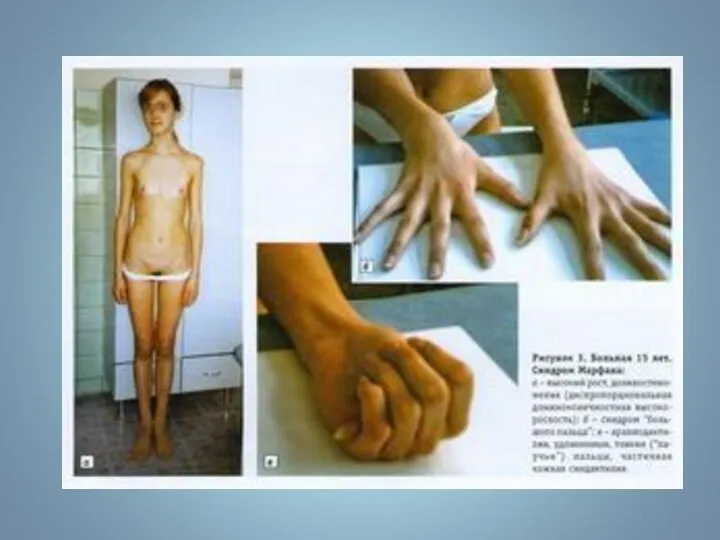
Симптом «большого пальца»
Арахнодактелия (длинные пальцы)
Симптом «запястья»
«Сандалевидная щель»
СИНДРОМ МАРФАНА
Симптом «большого пальца»
Арахнодактелия (длинные пальцы)
Симптом «запястья»
«Сандалевидная щель»
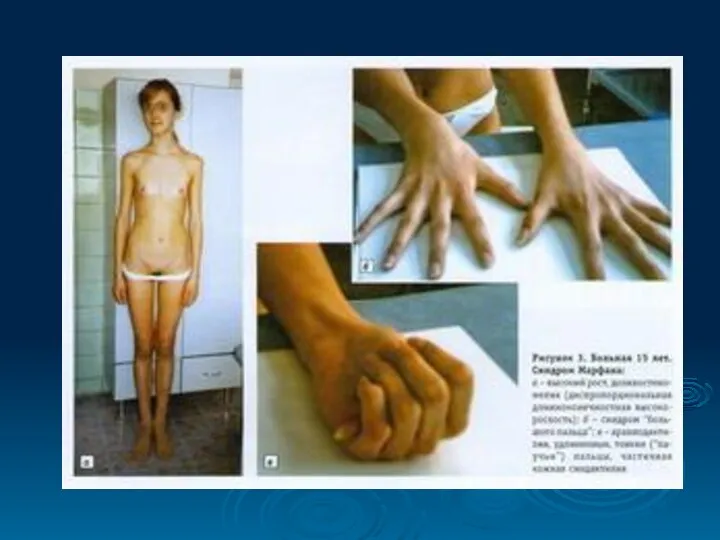
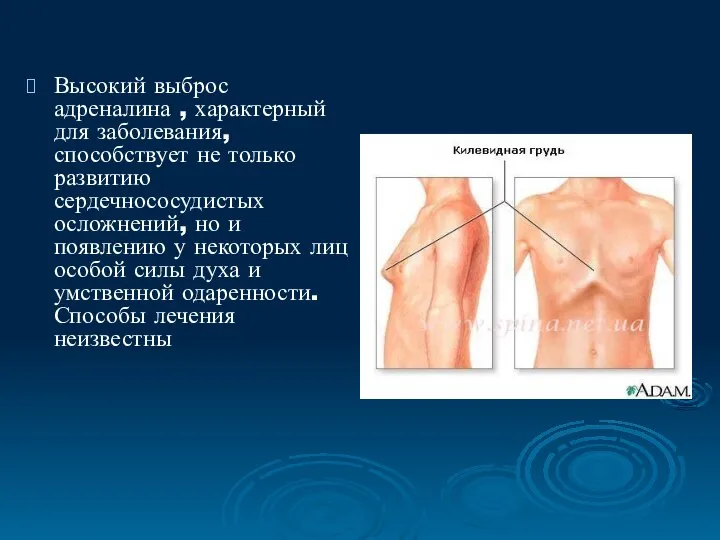
Высокий выброс адреналина , характерный для заболевания, способствует не только развитию
Высокий выброс адреналина , характерный для заболевания, способствует не только развитию
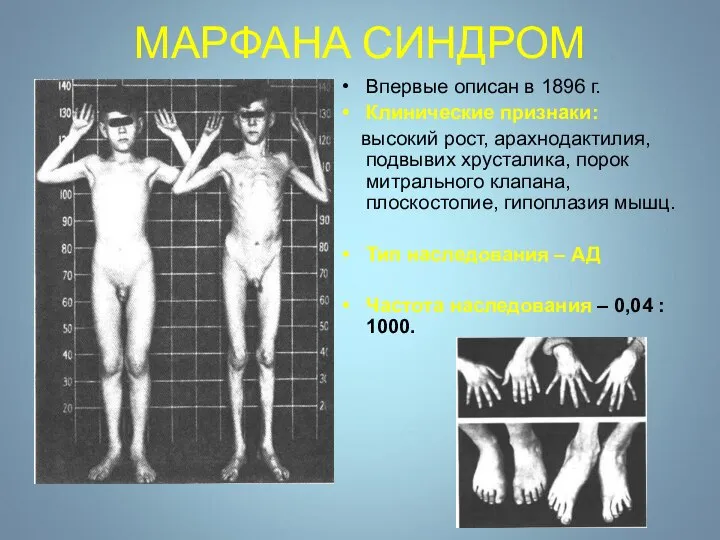
МАРФАНА СИНДРОМ
Впервые описан в 1896 г.
Клинические признаки:
высокий рост, арахнодактилия, подвывих
МАРФАНА СИНДРОМ
Впервые описан в 1896 г.
Клинические признаки:
высокий рост, арахнодактилия, подвывих
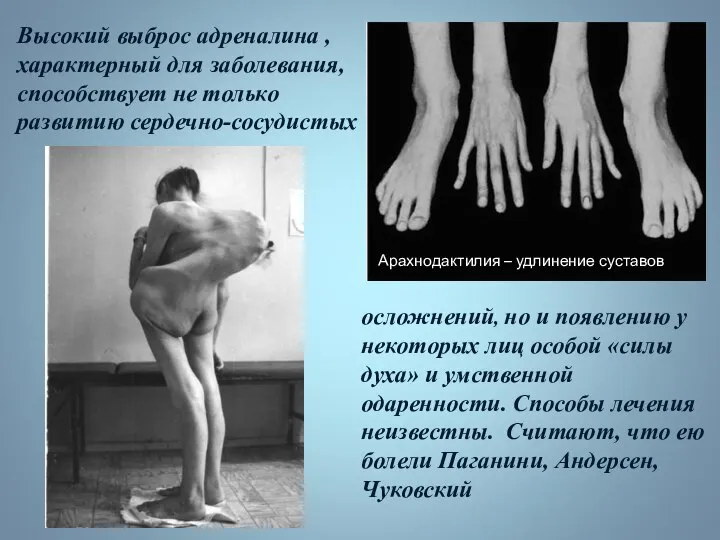
Высокий выброс адреналина , характерный для заболевания, способствует не только развитию
Высокий выброс адреналина , характерный для заболевания, способствует не только развитию
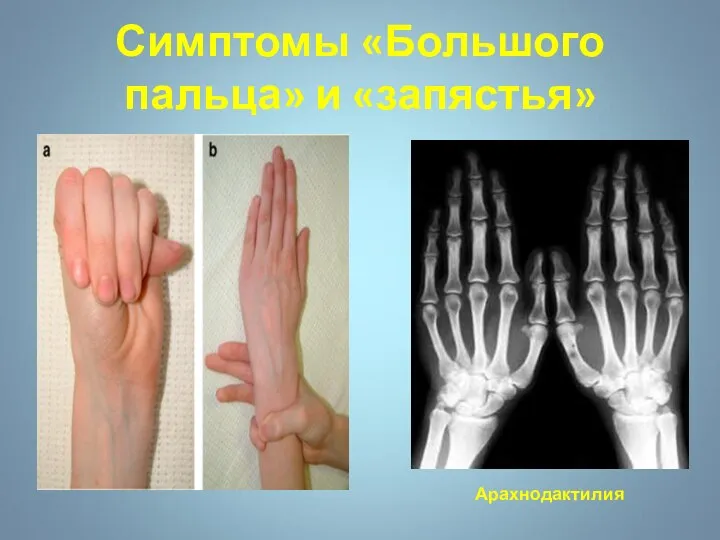
Симптомы «Большого пальца» и «запястья»
Арахнодактилия
Симптомы «Большого пальца» и «запястья»
Арахнодактилия
Синдром Марфана
Синдром Марфана

Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от
Синдром вызван наследственным пороком развития соединительной ткани. Больные часто умирают от

Известные люди с синдромом Марфана
Эхнатон, Паганини
Эхнатон, Паганини
Эхнатон Н. Паганини
Известные люди с синдромом Марфана
Эхнатон, Паганини
Эхнатон, Паганини
Эхнатон Н. Паганини

ФЕНИЛКЕТОНУРИЯ (ФЕНИЛПИРОВИНОГРАДНАЯ ОЛИГОФРЕНИЯ)
Фенилкетонурия (ФКУ) – это она из самых частых форм
ФЕНИЛКЕТОНУРИЯ (ФЕНИЛПИРОВИНОГРАДНАЯ ОЛИГОФРЕНИЯ)
Фенилкетонурия (ФКУ) – это она из самых частых форм

Классическая форма фенилкетонурии, ребенок в возрасте 1 год 7 мес.
Грубая задержка
Классическая форма фенилкетонурии, ребенок в возрасте 1 год 7 мес.
Грубая задержка
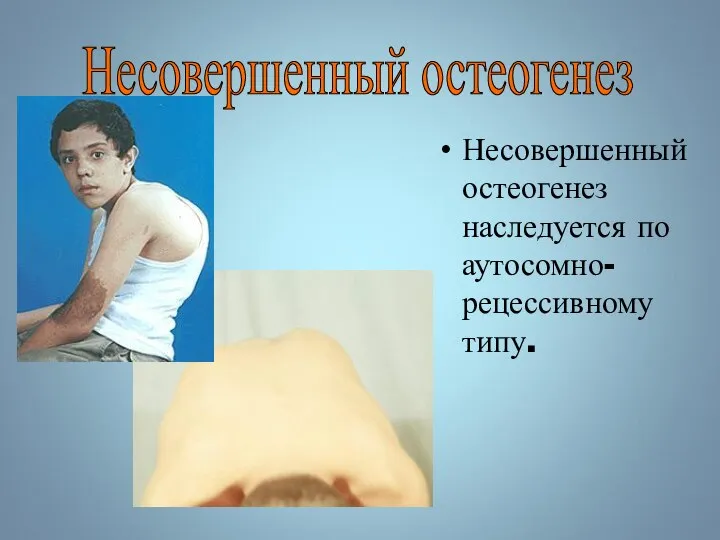
Несовершенный остеогенез наследуется по аутосомно-рецессивному типу.
Несовершенный остеогенез
Несовершенный остеогенез наследуется по аутосомно-рецессивному типу.
Несовершенный остеогенез

Несовершенный остеогенез фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями
Несовершенный остеогенез фенотипически проявляется повышенной ломкостью костей, вследствие нарушения остеогенеза, изменениями
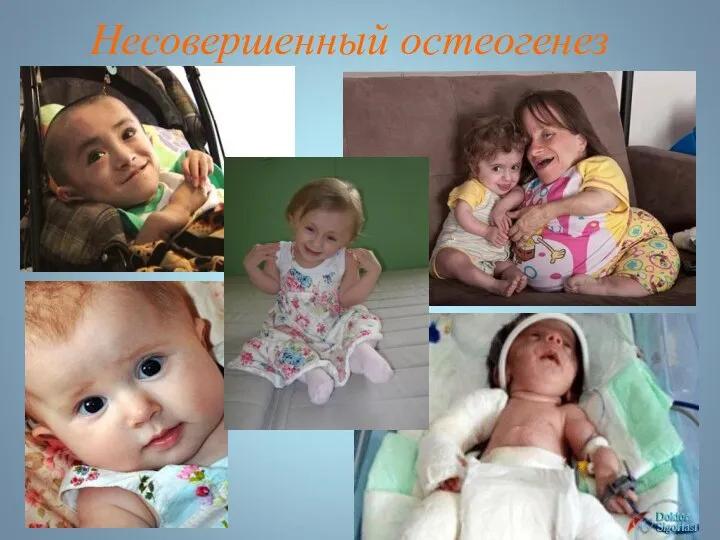
Несовершенный остеогенез
Несовершенный остеогенез

Ахондроплазия — врожденное поражение скелета — врождённая болезнь, характеризующаяся нарушением развития хрящевой
Ахондроплазия — врожденное поражение скелета — врождённая болезнь, характеризующаяся нарушением развития хрящевой

Человек, закончивших свой рост, достигает 30 - 41 см.
Человек, закончивших свой рост, достигает 30 - 41 см.

Причины мутации в настоящее время не известны.
Причины мутации в настоящее время не известны.

Знакомьтесь, это Джиоти Амге - 18-летняя студентка из Индии. Еще с
Знакомьтесь, это Джиоти Амге - 18-летняя студентка из Индии. Еще с
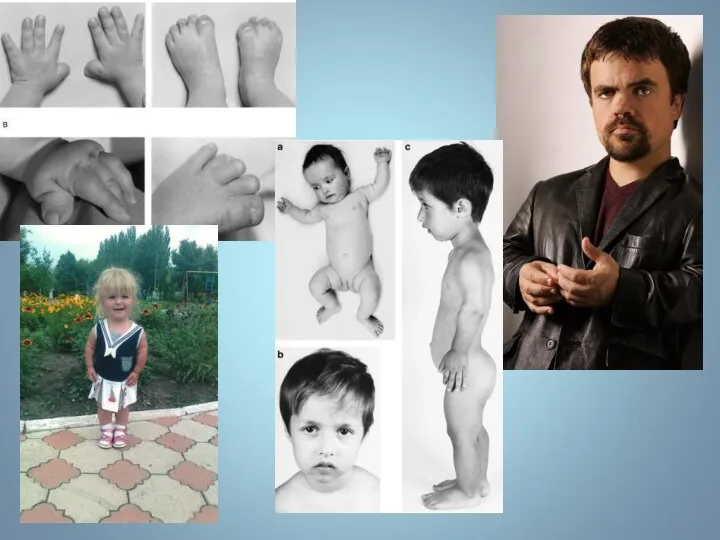
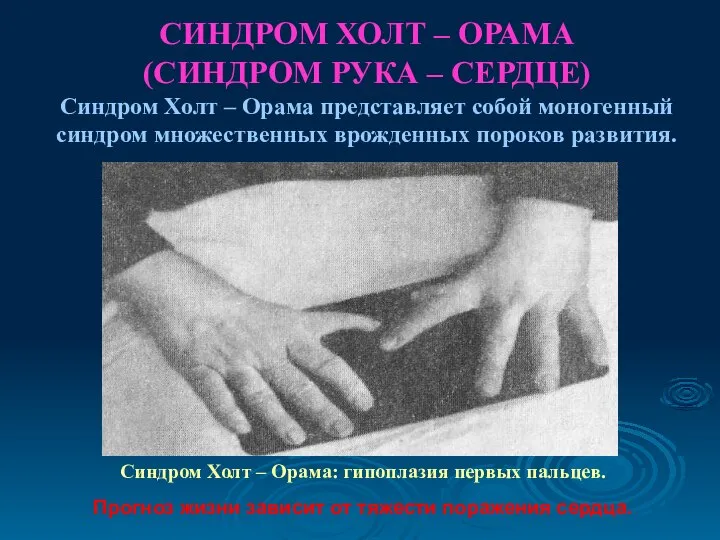
СИНДРОМ ХОЛТ – ОРАМА
(СИНДРОМ РУКА – СЕРДЦЕ)
Синдром Холт – Орама
СИНДРОМ ХОЛТ – ОРАМА (СИНДРОМ РУКА – СЕРДЦЕ) Синдром Холт – Орама
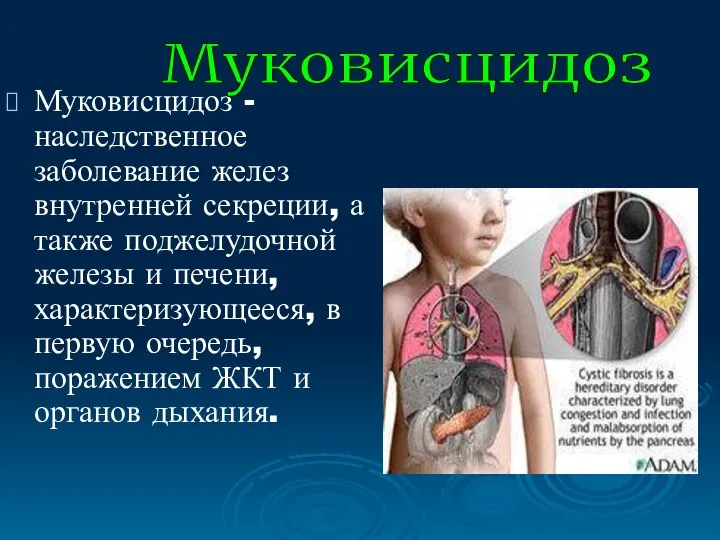
Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы
Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы

МУКОВИСЦИДОЗ (КИСТОФИБРОЗ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ)
Заболевание обусловлено генерализованным поражением экзокринных желез.
Частота муковисцедоза среди
МУКОВИСЦИДОЗ (КИСТОФИБРОЗ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ)
Заболевание обусловлено генерализованным поражением экзокринных желез.
Частота муковисцедоза среди
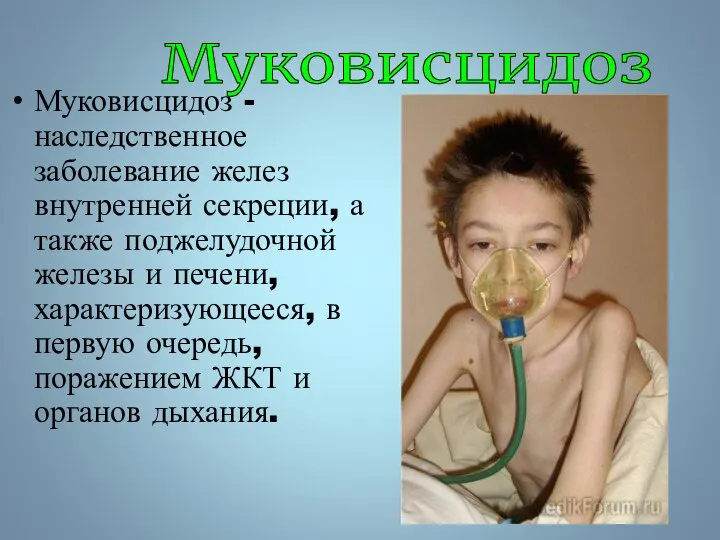
Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы
Муковисцидоз - наследственное заболевание желез внутренней секреции, а также поджелудочной железы
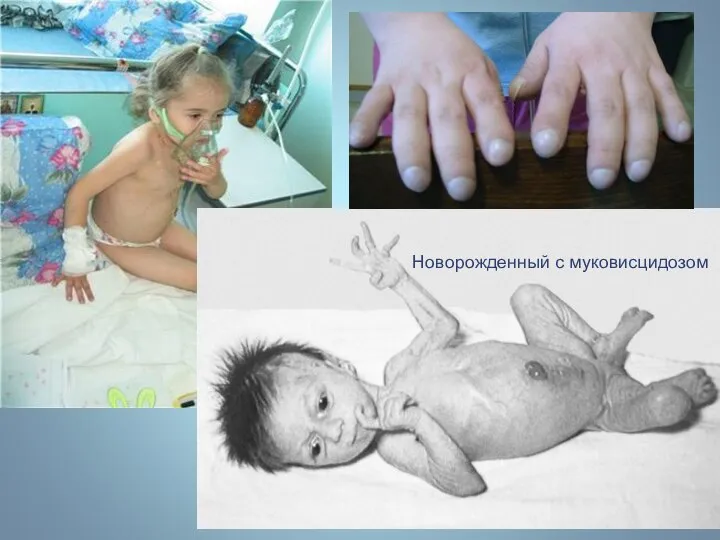
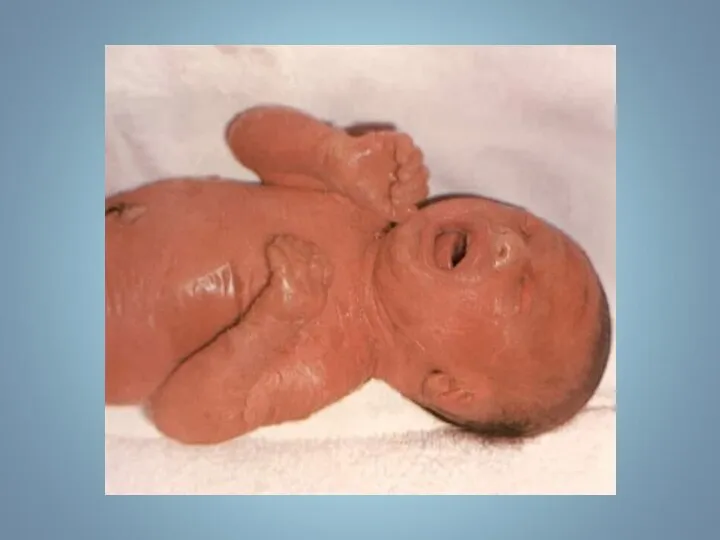
Новорожденный с муковисцидозом
Новорожденный с муковисцидозом
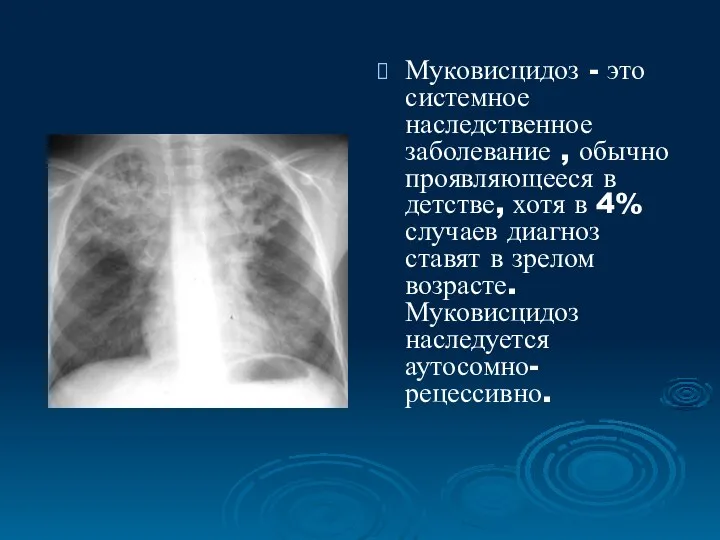
Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве,
Муковисцидоз - это системное наследственное заболевание , обычно проявляющееся в детстве,

Распространенность заболевания сильно отличается в разных этнических группах. Среди белого населения
Распространенность заболевания сильно отличается в разных этнических группах. Среди белого населения

Гомоцистинурия – нарушение метаболизма метионина.
При гомоцистинурии, как и при других наследственных
Гомоцистинурия – нарушение метаболизма метионина.
При гомоцистинурии, как и при других наследственных
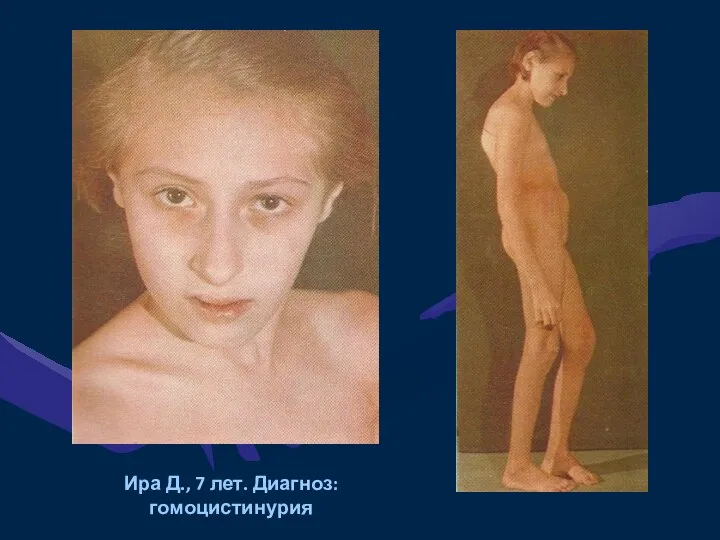
Ира Д., 7 лет. Диагноз: гомоцистинурия
Ира Д., 7 лет. Диагноз: гомоцистинурия
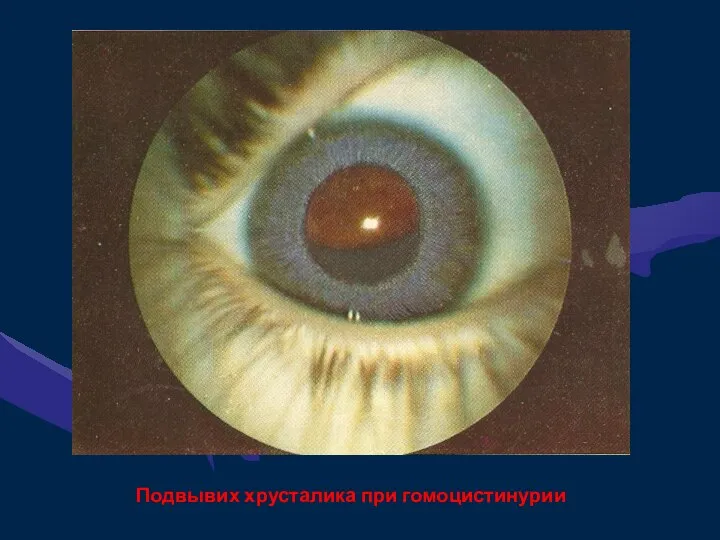
Подвывих хрусталика при гомоцистинурии
Подвывих хрусталика при гомоцистинурии

Материнская гистидинемия.
Нарушение обмена фермента гистидина
Описана в 1974 году Леоном у четверых
Материнская гистидинемия.
Нарушение обмена фермента гистидина
Описана в 1974 году Леоном у четверых
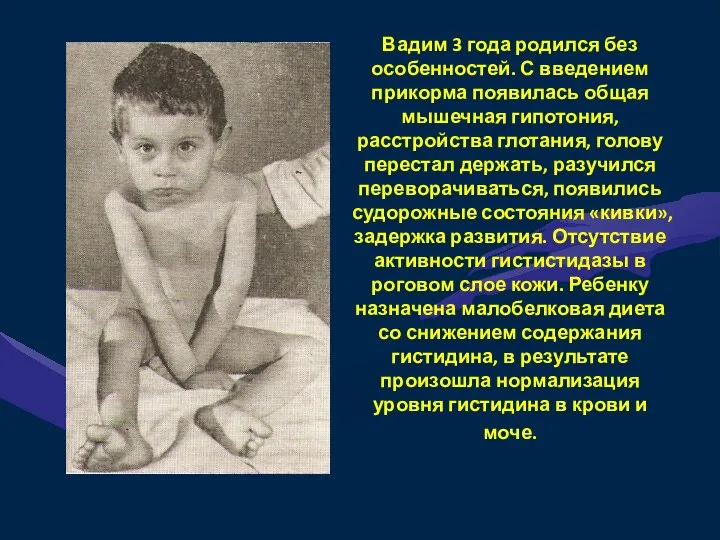
Вадим 3 года родился без особенностей. С введением прикорма появилась общая
Вадим 3 года родился без особенностей. С введением прикорма появилась общая

СИНДРОМ ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
Относится к редким заболеваниям, для которого характерен своеобразный симтомокомплекс –
СИНДРОМ ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
Относится к редким заболеваниям, для которого характерен своеобразный симтомокомплекс –
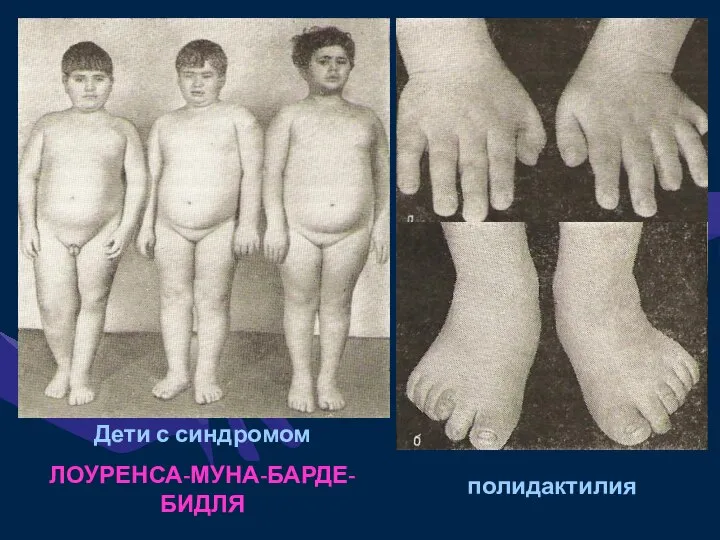
Дети с синдромом
ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
полидактилия
Дети с синдромом
ЛОУРЕНСА-МУНА-БАРДЕ-БИДЛЯ
полидактилия

Наследственные болезни обмена веществ соединительной ткани.
МУКОПОЛИСАХАРИДОЗЫ
Под термином «мукополисахаридозы» объединяется ряд патологических
Наследственные болезни обмена веществ соединительной ткани.
МУКОПОЛИСАХАРИДОЗЫ
Под термином «мукополисахаридозы» объединяется ряд патологических
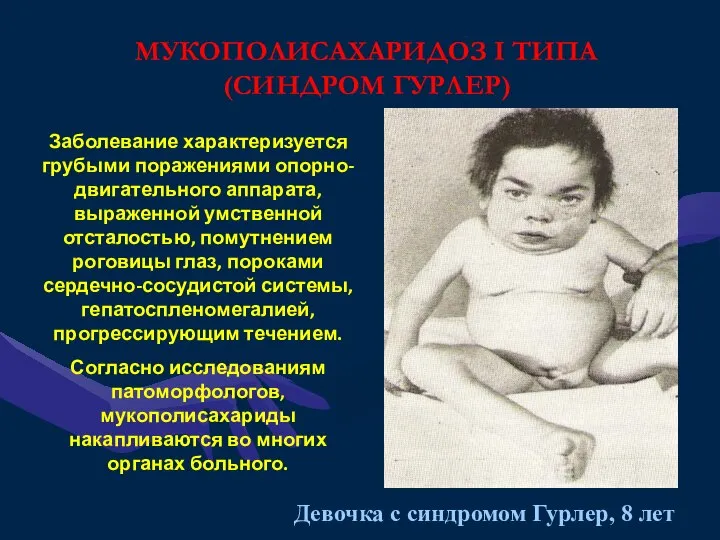
МУКОПОЛИСАХАРИДОЗ I ТИПА
(СИНДРОМ ГУРЛЕР)
Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной
МУКОПОЛИСАХАРИДОЗ I ТИПА
(СИНДРОМ ГУРЛЕР)
Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной
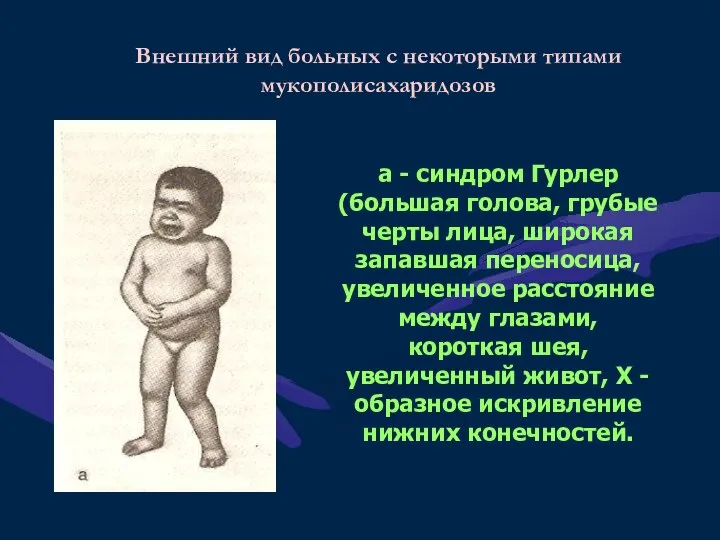
а - синдром Гурлер (большая голова, грубые черты лица, широкая запавшая
а - синдром Гурлер (большая голова, грубые черты лица, широкая запавшая
МУКОПОЛИСАХАРИДОЗ II ТИПА
(СИНДРОМ ГУНТЕРА)
В клинической картине на первый план выступает
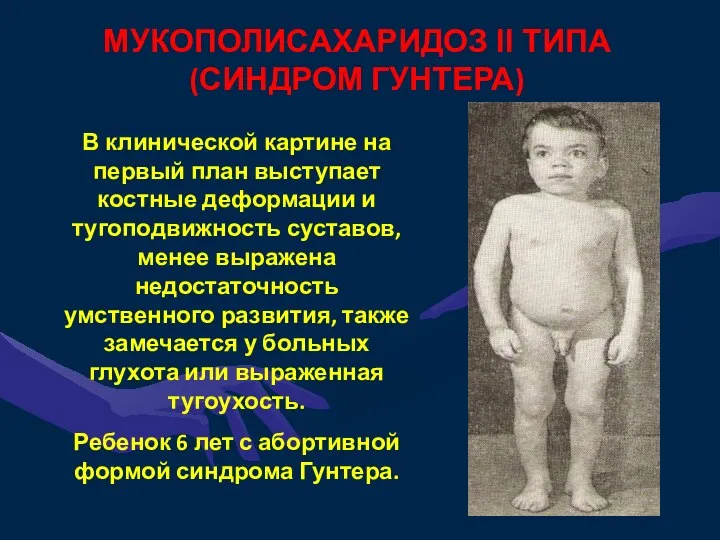
МУКОПОЛИСАХАРИДОЗ II ТИПА
(СИНДРОМ ГУНТЕРА)
В клинической картине на первый план выступает

МУКОПОЛИСАХАРИДОЗ III (А, В) ТИПА
(СИНДРОМ САНФИЛИППО)
МУКОПОЛИСАХАРИДОЗ IV ТИПА
(СИНДРОМ МОРКИО)
В
МУКОПОЛИСАХАРИДОЗ III (А, В) ТИПА
(СИНДРОМ САНФИЛИППО)
МУКОПОЛИСАХАРИДОЗ IV ТИПА
(СИНДРОМ МОРКИО)
В
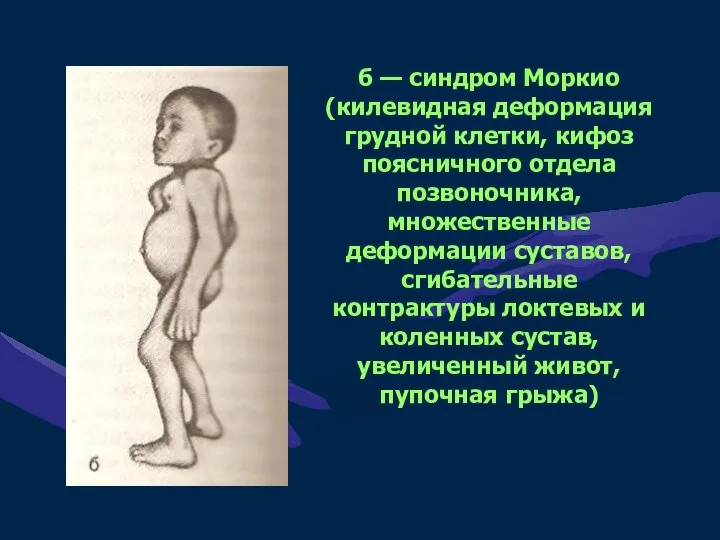
б — синдром Моркио (килевидная деформация грудной клетки, кифоз поясничного отдела
б — синдром Моркио (килевидная деформация грудной клетки, кифоз поясничного отдела
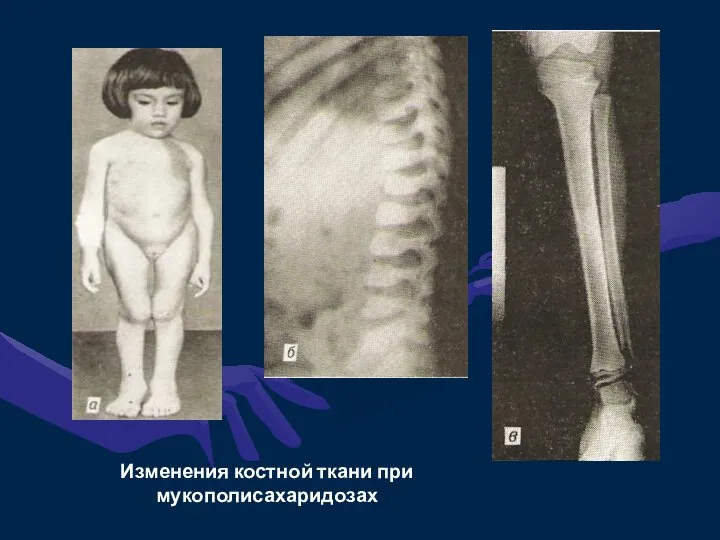
Изменения костной ткани при мукополисахаридозах
Изменения костной ткани при мукополисахаридозах

МУКОПОЛИСАХАРИДОЗ V ТИПА
(СИНДРОМ ШЕЙЕ)
МУКОПОЛИСАХАРИДОЗ VI ТИПА
(СИНДРОМ МАРОТО-ЛАМИ)
Клиническая картина заболевания
МУКОПОЛИСАХАРИДОЗ V ТИПА
(СИНДРОМ ШЕЙЕ)
МУКОПОЛИСАХАРИДОЗ VI ТИПА
(СИНДРОМ МАРОТО-ЛАМИ)
Клиническая картина заболевания
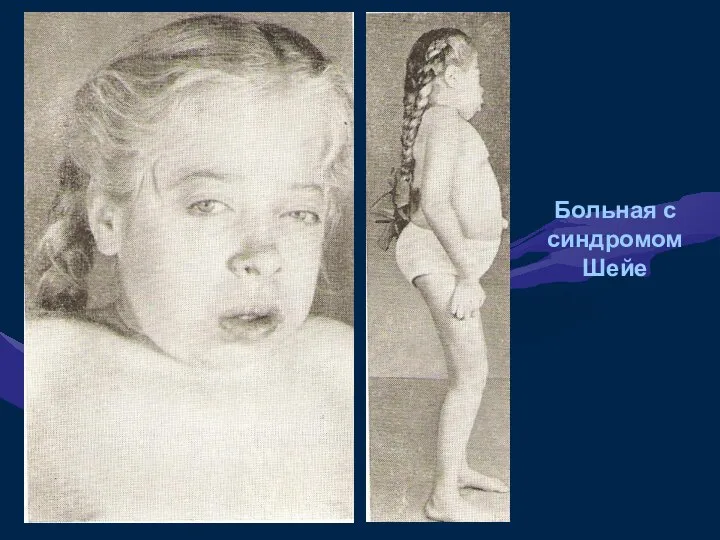
Больная с синдромом Шейе
Больная с синдромом Шейе
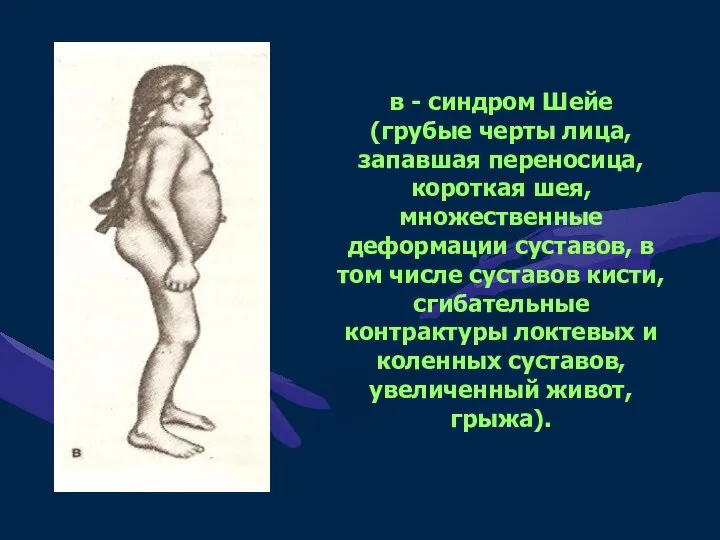
в - синдром Шейе (грубые черты лица, запавшая переносица, короткая шея,
в - синдром Шейе (грубые черты лица, запавшая переносица, короткая шея,
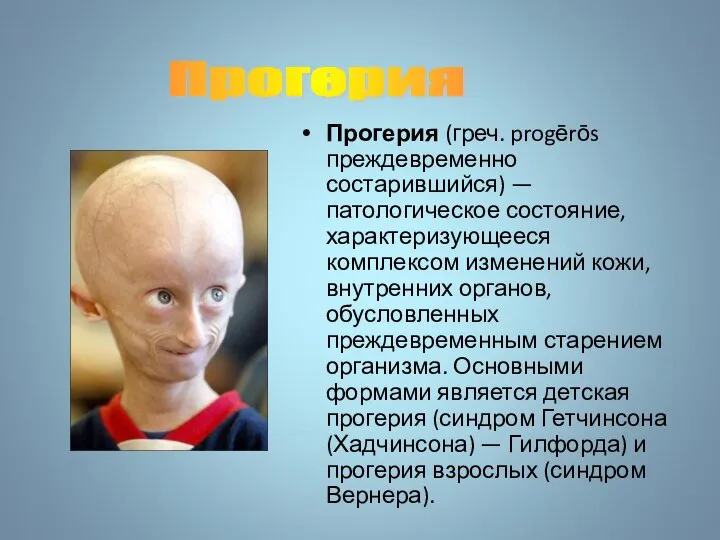
Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений
Прогерия (греч. progērōs преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений
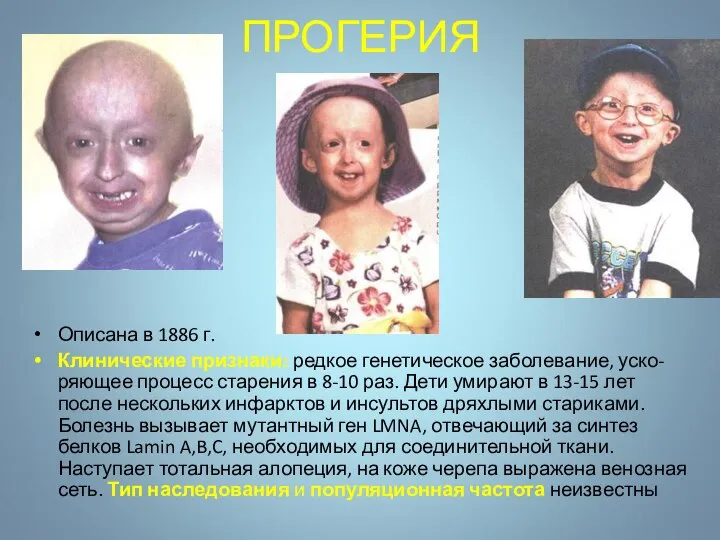
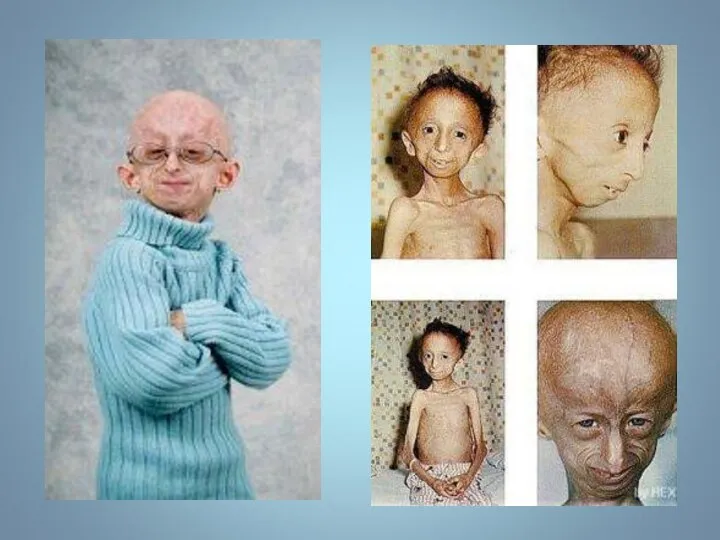
ПРОГЕРИЯ
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения
ПРОГЕРИЯ
Описана в 1886 г.
Клинические признаки: редкое генетическое заболевание, уско-ряющее процесс старения


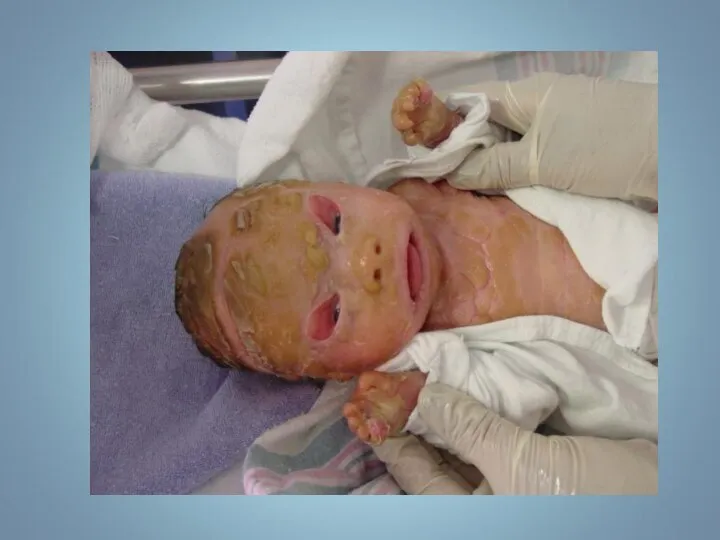
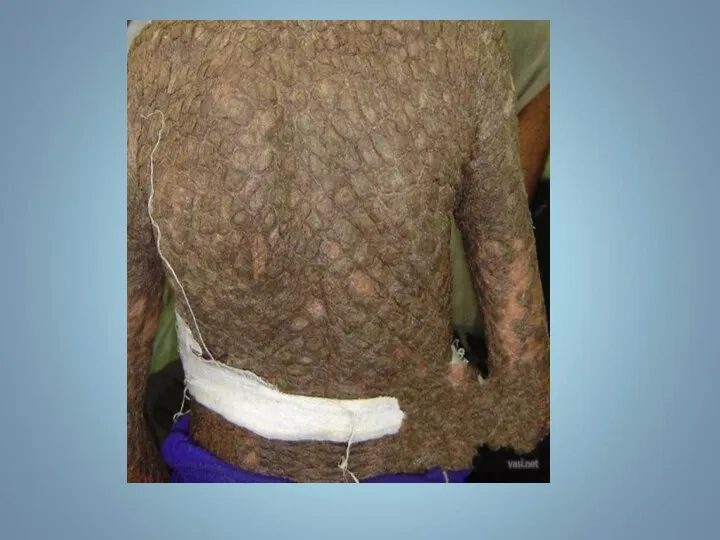
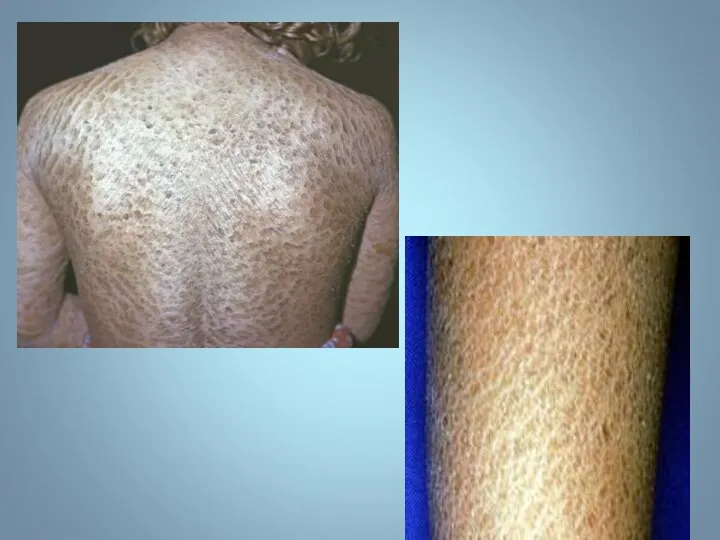
Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по
Ихтиоз (греч. - рыба) — наследственный дерматоз, характеризующийся диффузным нарушением ороговения по

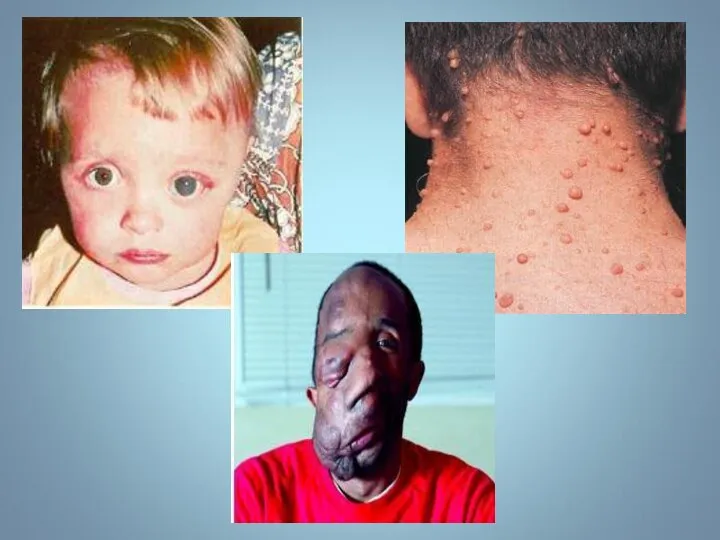
НЕЙРОФИБРОМАТОЗ
(БОЛЕЗНЬ РЕКЛИНГХАУЗЕНА)
Известно семь нозологических форм нейрофиброматоза (НФ), среди которых НФ
НЕЙРОФИБРОМАТОЗ (БОЛЕЗНЬ РЕКЛИНГХАУЗЕНА) Известно семь нозологических форм нейрофиброматоза (НФ), среди которых НФ
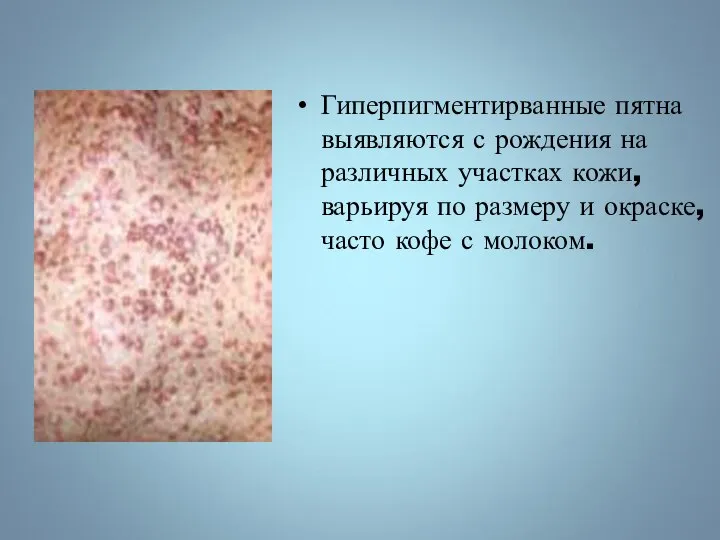
Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по
Гиперпигментирванные пятна выявляются с рождения на различных участках кожи, варьируя по

Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное
Чаще наблюдается нейрофиброматоз I типа ( болезнь Реклингхаузена ) - аутосомно-доминантное

Нейрофиброматоз
У больного этим заболеванием наблюдается: слоновость левой верхней конечности, обусловленная
Нейрофиброматоз
У больного этим заболеванием наблюдается: слоновость левой верхней конечности, обусловленная
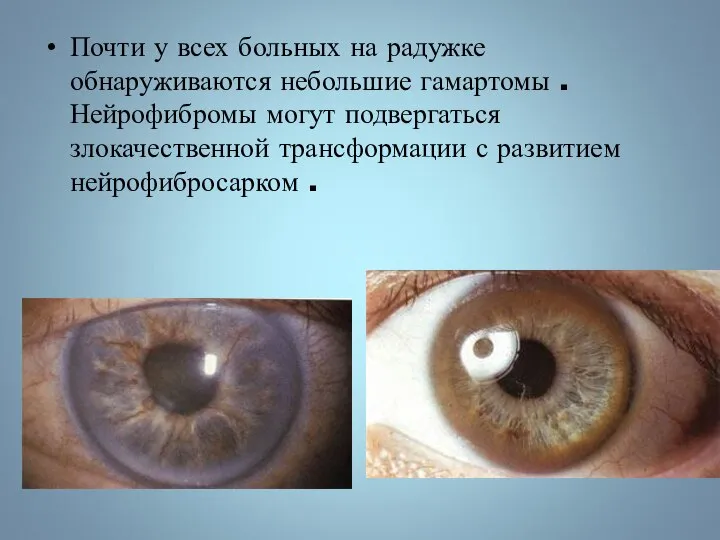
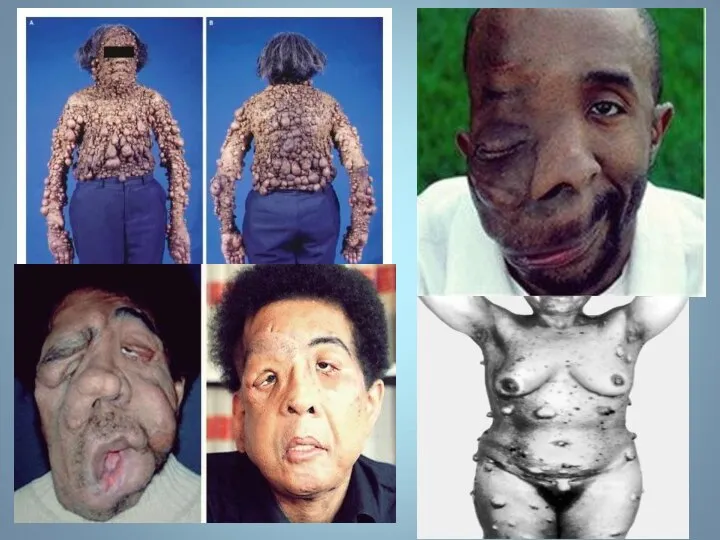
Почти у всех больных на радужке обнаруживаются небольшие гамартомы . Нейрофибромы
Почти у всех больных на радужке обнаруживаются небольшие гамартомы . Нейрофибромы

Мутация, за развитие этого типа, обнаружена в хромосоме 17, в зоне,
Мутация, за развитие этого типа, обнаружена в хромосоме 17, в зоне,
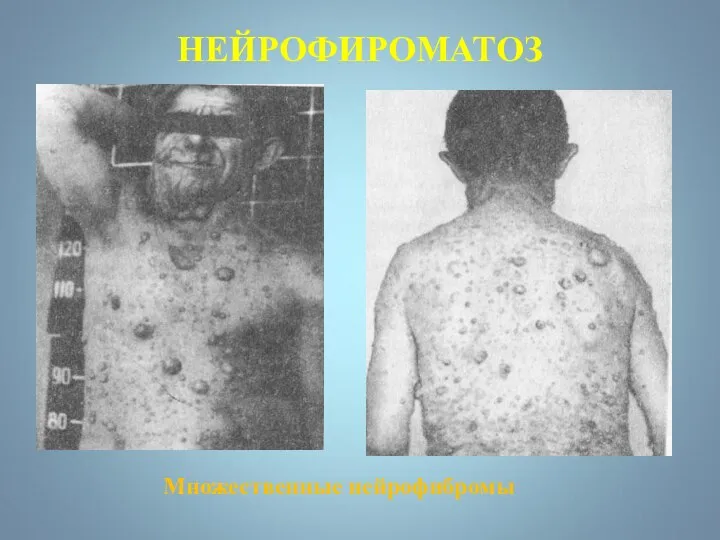
НЕЙРОФИРОМАТОЗ
Множественные нейрофибромы
НЕЙРОФИРОМАТОЗ
Множественные нейрофибромы


АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (ВРАЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ)
Адреногенитальный синдром (АГС) относится к группе
АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ (ВРАЖДЕННАЯ ГИПЕРПЛАЗИЯ КОРЫ НАДПОЧЕЧНИКОВ)
Адреногенитальный синдром (АГС) относится к группе
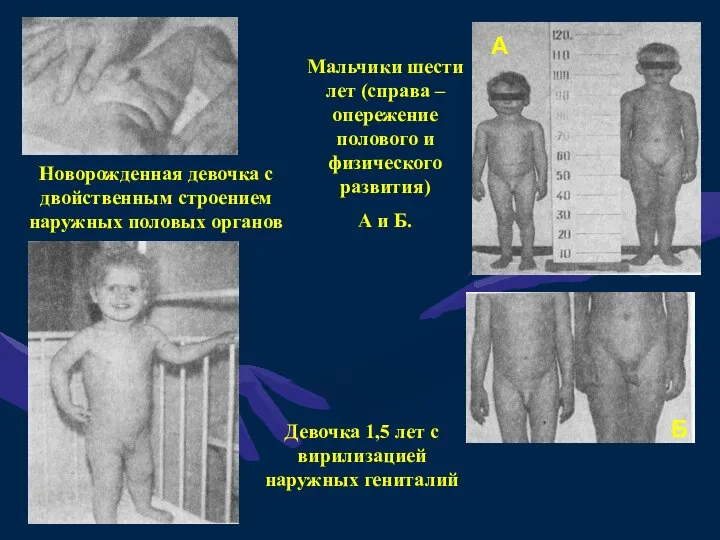
Новорожденная девочка с двойственным строением наружных половых органов
Мальчики шести лет (справа
Новорожденная девочка с двойственным строением наружных половых органов
Мальчики шести лет (справа


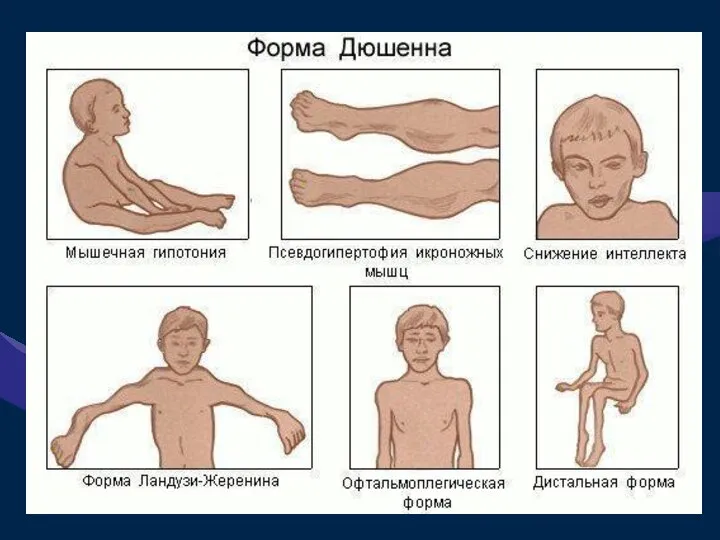
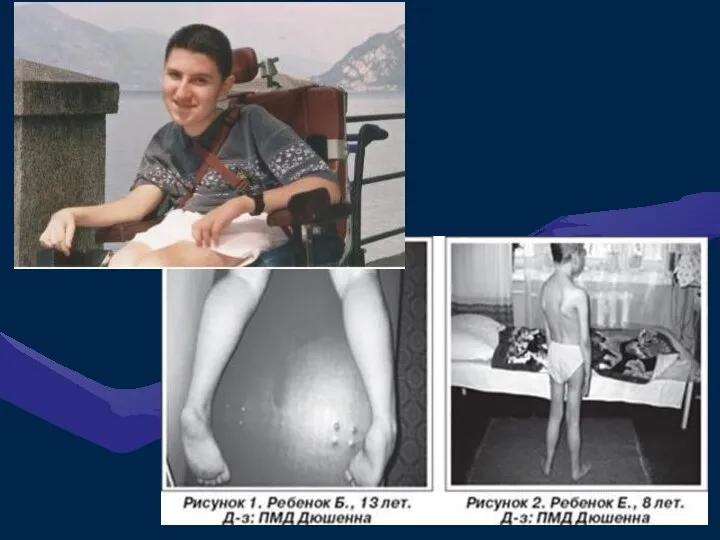
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ДЮШЕННА)
Это одна из самых частых форм наследственных нервно
ПСЕВДОГИПЕРТРОФИЧЕСКАЯ МЫШЕЧНАЯ ДИСТРОФИЯ (ДЮШЕННА)
Это одна из самых частых форм наследственных нервно
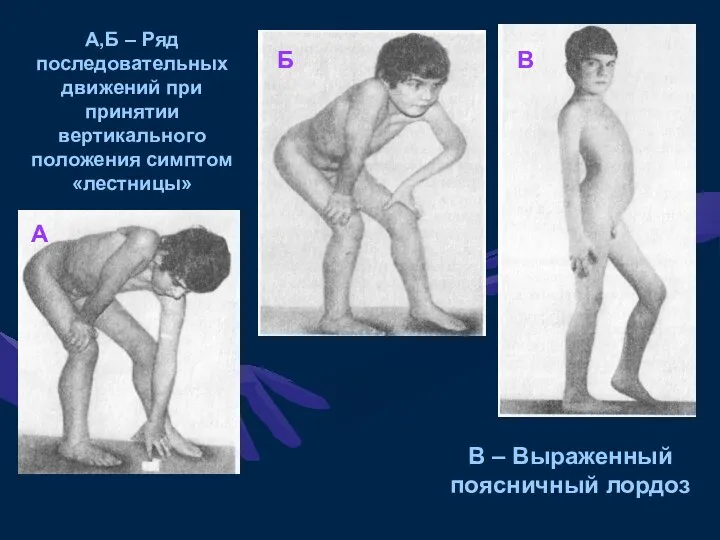
А,Б – Ряд последовательных движений при принятии вертикального положения симптом «лестницы»
А
Б
В
В
А,Б – Ряд последовательных движений при принятии вертикального положения симптом «лестницы»
А
Б
В
В

Странное племя людей-страусов (сапади) в Центральной Африке отличает от прочих обитателей
Странное племя людей-страусов (сапади) в Центральной Африке отличает от прочих обитателей

Синдром Ангельмана
Синдром Ангельмана (СА) - это нейро-генетическое заболевание, характеризующееся интеллектуальной и физической задержкой
Синдром Ангельмана
Синдром Ангельмана (СА) - это нейро-генетическое заболевание, характеризующееся интеллектуальной и физической задержкой



Дети наркоманов. Копия, воск.
Дети наркоманов. Копия, воск.
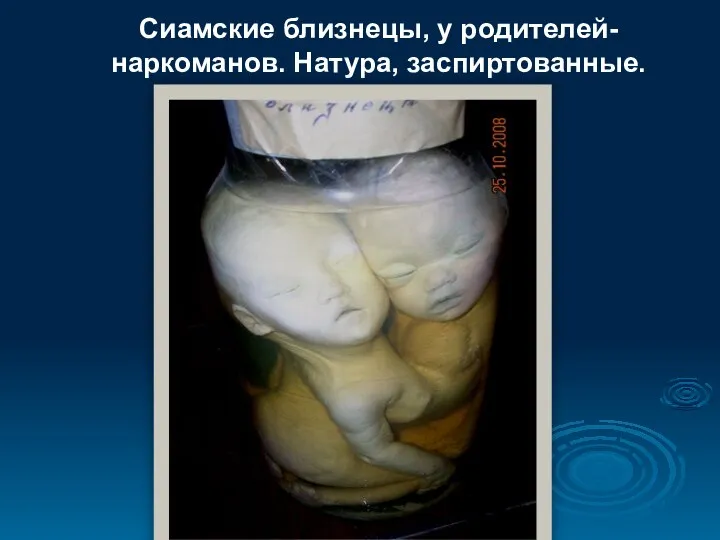
Сиамские близнецы, у родителей-наркоманов. Натура, заспиртованные.
Сиамские близнецы, у родителей-наркоманов. Натура, заспиртованные.

Дети у родителей больных наследственными заболеваниями. Копия, воск.
Дети у родителей больных наследственными заболеваниями. Копия, воск.
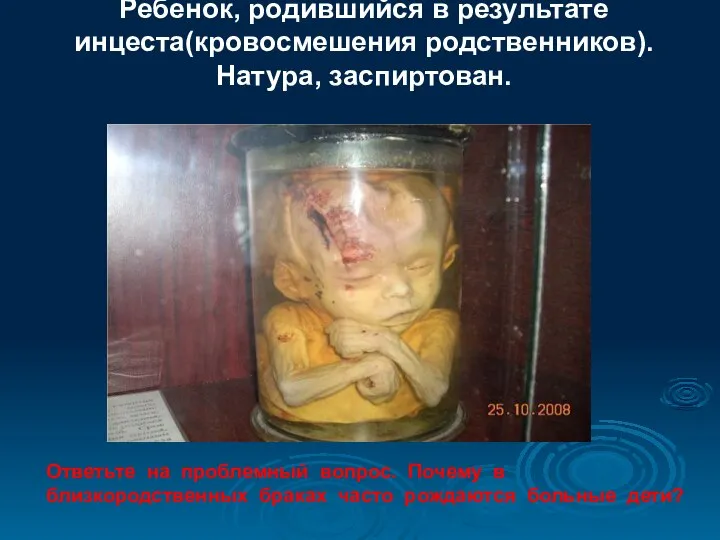
Ребенок, родившийся в результате
инцеста(кровосмешения родственников).
Натура, заспиртован.
Ответьте на проблемный вопрос.
Ребенок, родившийся в результате
инцеста(кровосмешения родственников).
Натура, заспиртован.
Ответьте на проблемный вопрос.

Ребенок, родившийся
в семье чернобыльцев. Натура, мумия.
Ребенок, родившийся
в семье чернобыльцев. Натура, мумия.

Человек-циклоп, и женщина-слон.
Жили в 19 веке. Копия, воск.
Человек-циклоп, и женщина-слон.
Жили в 19 веке. Копия, воск.
Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом,
Согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом,

Если принять, что у человека примерно
100 000 генов и
Если принять, что у человека примерно
100 000 генов и

Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации
Вся история развития человека есть непрерывная цепь мутаций в нем. Мутации
 Презентация на тему "Костные рыбы" - скачать бесплатно презентации по Биологии
Презентация на тему "Костные рыбы" - скачать бесплатно презентации по Биологии Белки и нуклеиновые кислоты
Белки и нуклеиновые кислоты Надотдел Моховидные
Надотдел Моховидные Институт генетики и цитологии Национальной академии наук Беларуси
Институт генетики и цитологии Национальной академии наук Беларуси Генетика и селекция. Индивидуальный отбор
Генетика и селекция. Индивидуальный отбор Дикие животные (животные наших лесов)
Дикие животные (животные наших лесов) Сравнительная характеристика голосеменных и покрытосеменных растений
Сравнительная характеристика голосеменных и покрытосеменных растений Хищные Совы. Falconiformes
Хищные Совы. Falconiformes Растениеводство, как наука и отрасль
Растениеводство, как наука и отрасль Фотосинтез. Открытие процесса фотосинтеза
Фотосинтез. Открытие процесса фотосинтеза Клітинне дихання. Біохімічні механізми дихання
Клітинне дихання. Біохімічні механізми дихання Видоизмененные побеги
Видоизмененные побеги Презентация на тему Пищеварение в ротовой полости
Презентация на тему Пищеварение в ротовой полости Цитология – наука, изучающая клетку. Многообразие клеток Подготовила учитель биологии МБОУ СОШ с. Виляйки Свищева Л.А.
Цитология – наука, изучающая клетку. Многообразие клеток Подготовила учитель биологии МБОУ СОШ с. Виляйки Свищева Л.А. Возникновение и развитие жизни на земле
Возникновение и развитие жизни на земле Онтофилогенетические закономерности развития эволюции систем органов
Онтофилогенетические закономерности развития эволюции систем органов Внешнее строение и скелет костных рыб
Внешнее строение и скелет костных рыб Пластический обмен в клетке. Биосинтез белка в клетке. Генетический код
Пластический обмен в клетке. Биосинтез белка в клетке. Генетический код Презентация на тему "Исследование рекреационных нагрузок в лесах, предназначенных для отдыха населения" - скачать презентац
Презентация на тему "Исследование рекреационных нагрузок в лесах, предназначенных для отдыха населения" - скачать презентац Что такое нейронные сети?
Что такое нейронные сети? Добрая зима. Перелётные птицы
Добрая зима. Перелётные птицы Водно-минеральный обмен
Водно-минеральный обмен Вирусы. Систематическое положение вирусов в системе органического мира
Вирусы. Систематическое положение вирусов в системе органического мира Эмбриональное развитие человека
Эмбриональное развитие человека Улы саңырауқұлақтар
Улы саңырауқұлақтар Нервная и гуморальная регуляция функций организма
Нервная и гуморальная регуляция функций организма Французкая линька
Французкая линька Аттестационная работа. Проектная деятельность учащихся 8-9 классов по биологии, формирование метапредметных результатов обучения
Аттестационная работа. Проектная деятельность учащихся 8-9 классов по биологии, формирование метапредметных результатов обучения