- Наследственно-дегенеративные заболевания нервной системы

Содержание
- 2. Наследственно-дегенеративные заболевания нервной системы. Доцент: Боброва Л.В.
- 3. План лекции: Общая характеристика заболеваний. Классификация. Патогенез и патоморфология. Клиническая картина, примеры, фотографии больных. Дополнительные методы
- 4. Наследственно-дегенеративные заболевания нервной системы – обширная группа болезней, обусловленных изменениями генетической информации (генные мутации). Генные мутации
- 5. Классификация: I. Миопатии: 1. Мышечная дистрофия Дюшенна. 2. Миопатия Эрба (конечностно-поясная форма). 3. Лице-лопаточно-плечевая форма Ландузи-Дежерина.
- 6. II.а Спинальные амиотрофии: 1. Спинальная амиотрофия Вердинга-Гоффманна. 2. Прогрессирующая спинальная амиотрофия Кугельберга-Веландера. II.б Невральные амиотрофии. Невральная
- 7. III. Миастения. IV. Миатония. Врожденная миатония (Болезнь Оппенгейма). V. Миотония Томсона. VI. Пароксизмальная миоплегия.
- 8. Патоморфология миопатий: Нарушение распределения типов мышечных волокон. Изменение размера мышечных волокон. Нарушение строения мышечных волокон. Патологические
- 9. Патоморфология амиотрофий: Грубое поражение клеток передних рогов, передних корешков и периферических нервов спинного мозга: уменьшение количества
- 10. Патоморфология миастении: 1. Нарушение синаптического проведения импульсов. 2. Эндокринные расстройства, особенно дисфункция вилочковой железы. 3. В
- 11. Патоморфология миатонии: Недоразвитие клеток передних рогов спинного мозга. Дегенеративные изменения мышечных волокон.
- 12. Патогенез пароксизмальной миоплегии: Нарушение синаптической передачи и недостаточная генерация потенциала концевых пластинок. Нарушение распространения потенциала действия
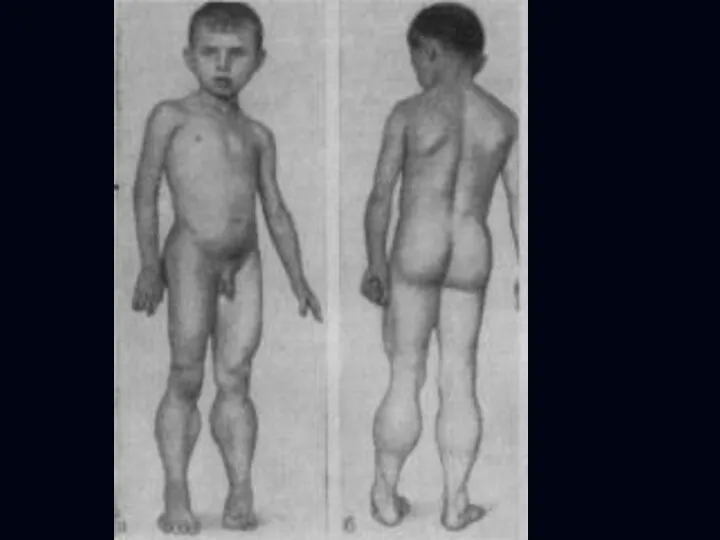
- 13. Мышечная дистрофия Дюшенна: Начало заболевания в раннем возрасте – 3 – 4 года. Преимущественно поражение мышц
- 14. Сухожильные рефлексы вначале снижаются, затем исчезают. Отмечаются атрофия мышц языка, мягкого нёба, гортани и жевательных мышц.
- 17. Юношеская миопатия Эрба-Рота: Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей). Болезнь начинается в возрасте от
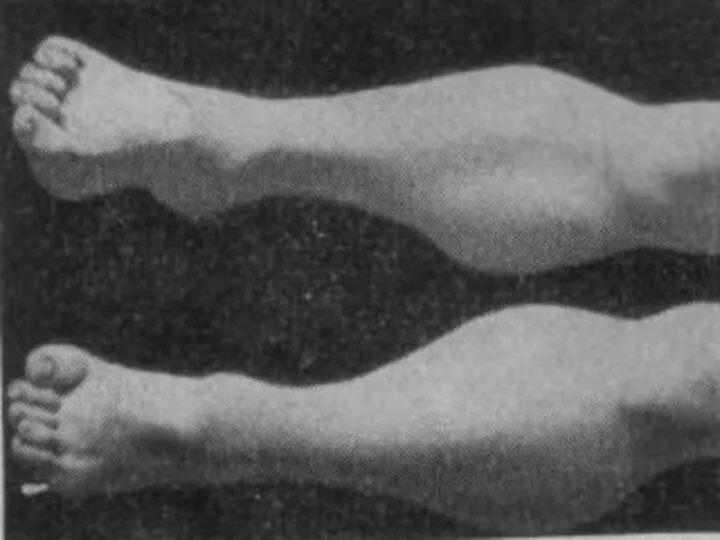
- 18. Больные испытывают затруднение при вставании с пола, помогая себе при этом руками; по ступенькам лестницы поднимаются,
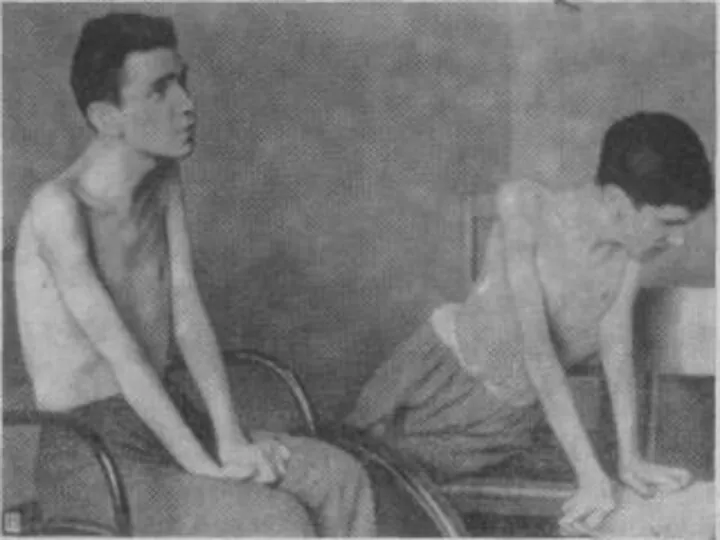
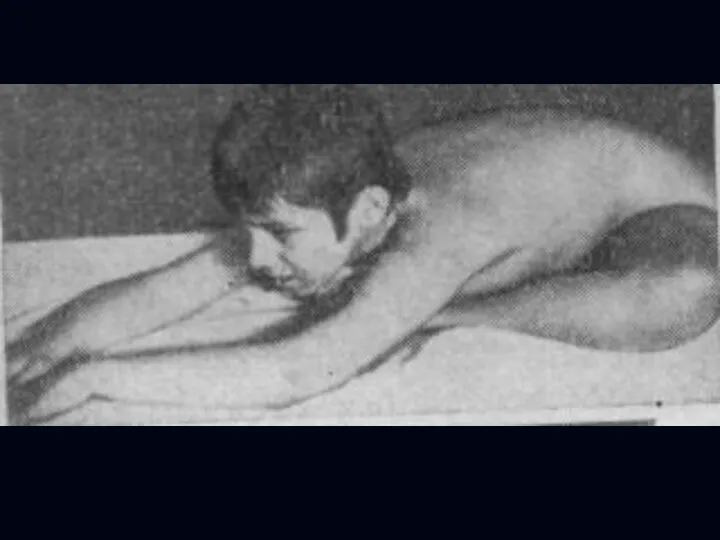
- 27. Лице-лопаточно-плечевая форма Ландузи-Дежерина. Начало заболевания с 7 - 15 лет. Слабость и атрофии мышц лица и
- 28. Симптом «крыловидных лопаток», горизонтальное расположение ключиц. Характерны толстые, выпяченные вперед губы («губы тапира»), «поперечная улыбка». Выраженный
- 30. Спинальная амиотрофия Вердига – Гоффмана. Выделяют формы: Врожденная форма. Ранняя детская форма. Поздняя форма.
- 31. Врожденная форма: Начало внутриутробно. Слабое шевеление плода. При рождении вялый паралич в проксимальных отделах конечностей. Бульбарные
- 32. Ранняя детская форма: Начало до 1,5 лет. После интеркурретного заболевания ребенок начинает терять приобретенные навыки. Вялые
- 33. Поздняя форма: Начало в 1,5 – 2 года. Вялые параличи проксимальных отделов ног, затем рук. Мышечные
- 38. Спинальная амиотрофия Кугельберга-Веландера (псевдомиопатическая). Тип наследования аутосомно-рецессивный. Начало в 6 лет. Течение доброкачественное, ещё в 30
- 39. Невральная амиотрофия Шарко-Мари Тип наследования аутосомно-доминантный. Начальные проявления с 13-17 лет. Характерны дистальные парезы, атрофии, сухожильная
- 40. Атрофия мышц голеней и нижней трети бедер. Ноги по виду напоминают «ноги аиста». Часто сочетается с
- 42. Миастения: Появляется утомляемость, распространяется на мышцы: губ, жевательные, глазодвигательные, глотательные. Наблюдается птоз, диплопия, поперечная улыбка (симптом
- 43. Утомляемость появляется в мышцах конечностей. Наблюдается увеличение вилочковой железы. Заболевание имеет тенденцию к прогрессированию. Могут возникать
- 44. Врожденная миатония Оппенгейма: С рождения – мало активных движений. Резкая гипотония или полная атония мышц. Отмечается
- 45. Отсутствуют также гипертрофия и псевдогипертрофия. Лицевые мышцы поражаются редко. Психика сохранена. Тазовых и сенсорных нарушений нет.
- 52. Пароксизмальная миоплегия - генетически детерминированные нервно-мышечные заболевания, обусловленные нарушениями обмена калия и характеризующиеся приступами вялого паралича
- 53. Гипокалиемическая форма: 1. Начало от 10 до 16 лет. 2. Возникновение приступов часто связано с циклом
- 54. 6. При парциальном приступе мышечная слабость и парез или плегия развивается на одной конечности, реже в
- 55. Гиперкалиемическая форма: 1. Начало от 10 до 18 лет. 2. Возникают приступы пароксизмаль-ного гиперкалиемического паралича, как
- 56. 5. В последующем развивается мышечная слабость, гипотония мышц, снижение сухожильных рефлексов. 6. Приступ сопровождается выраженными вегетативными
- 57. Нормокалиемическая форма: 1. Начало в первые 10 лет жизни. 2. Сочетание признаков мышечной слабости и ограничения
- 58. Дополнительные методы обследования: Электромиография (ЭМГ). Биопсия скелетной мышцы. Биохимический анализ крови (БАК). Компьютерная томография скелетных мышц.
- 59. Лечение прогрессирующих мышечных миопатий: Для улучшения трофики: АТФ. Антихолинэстеразные препараты: галантамин. Витаминотерапия: витамины группы В, Е.
- 60. Лечение прогрессирующих мышечных амиотрофий: Для улучшения трофики: АТФ, глютаминовая кислота, метионин, лейцин. Антихолинэстеразные препараты: прозерин, галантамин,
- 61. Лечение невральной амиотрофии Шарко-Мари: Антихолинэстеразные препараты: прозерин, дибазол. Для улучшения трофики: АТФ. ЛФК, массаж, четырехкамерные ванны,
- 62. Лечение миастении: Антихолинэстеразные препараты: прозерин, местинон, оксазил. Препарат, увеличивающий нервно-мышечную передачу – хлористый калий. Рентгенотерапия на
- 63. Лечение пароксизмальной миоплегии: Во время приступа. Высокие дозы хлорида калия внутрь (10-15 г в виде раствора)
- 64. В межприступный период: 1) Диета с высоким содержанием калия и ограничением углеводов и натрия. 2) Спиронолактон,
- 65. Литература: 1. «Детская неврология», Л.О. Бадалян, 2003г. 2. «Наследственные болезни нервной системы» Ю.Е. Вельтищева, П.А. Темина,
- 66. 4. «Неврология детского возраста», А.С. Петрухин, 2004г. 5. «Нервные болезни», С.И. Гусев, 2005г.
- 68. Скачать презентацию

Наследственно-дегенеративные заболевания нервной системы.
Доцент: Боброва Л.В.
Наследственно-дегенеративные заболевания нервной системы.
Доцент: Боброва Л.В.

План лекции:
Общая характеристика заболеваний.
Классификация.
Патогенез и патоморфология.
Клиническая картина, примеры, фотографии больных.
Дополнительные методы
План лекции:
Общая характеристика заболеваний.
Классификация.
Патогенез и патоморфология.
Клиническая картина, примеры, фотографии больных.
Дополнительные методы

Наследственно-дегенеративные заболевания нервной системы
– обширная группа болезней, обусловленных изменениями генетической
Наследственно-дегенеративные заболевания нервной системы – обширная группа болезней, обусловленных изменениями генетической

Классификация:
I. Миопатии:
1. Мышечная дистрофия Дюшенна.
2. Миопатия Эрба (конечностно-поясная форма).
Классификация:
I. Миопатии:
1. Мышечная дистрофия Дюшенна.
2. Миопатия Эрба (конечностно-поясная форма).

II.а Спинальные амиотрофии:
1. Спинальная амиотрофия Вердинга-Гоффманна.
2. Прогрессирующая спинальная
II.а Спинальные амиотрофии:
1. Спинальная амиотрофия Вердинга-Гоффманна.
2. Прогрессирующая спинальная

III. Миастения.
IV. Миатония.
Врожденная миатония
(Болезнь Оппенгейма).
V. Миотония Томсона.
VI. Пароксизмальная миоплегия.
III. Миастения.
IV. Миатония.
Врожденная миатония
(Болезнь Оппенгейма).
V. Миотония Томсона.
VI. Пароксизмальная миоплегия.

Патоморфология миопатий:
Нарушение распределения типов мышечных волокон.
Изменение размера мышечных волокон.
Нарушение строения мышечных
Патоморфология миопатий:
Нарушение распределения типов мышечных волокон.
Изменение размера мышечных волокон.
Нарушение строения мышечных

Патоморфология амиотрофий:
Грубое поражение клеток передних рогов, передних корешков и
Патоморфология амиотрофий:
Грубое поражение клеток передних рогов, передних корешков и

Патоморфология миастении:
1. Нарушение синаптического проведения импульсов.
2. Эндокринные расстройства, особенно дисфункция вилочковой
Патоморфология миастении:
1. Нарушение синаптического проведения импульсов.
2. Эндокринные расстройства, особенно дисфункция вилочковой

Патоморфология миатонии:
Недоразвитие клеток передних рогов спинного мозга.
Дегенеративные изменения мышечных волокон.
Патоморфология миатонии:
Недоразвитие клеток передних рогов спинного мозга.
Дегенеративные изменения мышечных волокон.

Патогенез пароксизмальной миоплегии:
Нарушение синаптической передачи и недостаточная генерация потенциала концевых пластинок.
Нарушение
Патогенез пароксизмальной миоплегии:
Нарушение синаптической передачи и недостаточная генерация потенциала концевых пластинок.
Нарушение

Мышечная дистрофия Дюшенна:
Начало заболевания в раннем возрасте – 3 – 4
Мышечная дистрофия Дюшенна:
Начало заболевания в раннем возрасте – 3 – 4

Сухожильные рефлексы вначале снижаются, затем исчезают.
Отмечаются атрофия мышц языка, мягкого нёба,
Отмечаются атрофия мышц языка, мягкого нёба,

Юношеская миопатия Эрба-Рота:
Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей).
Болезнь
Юношеская миопатия Эрба-Рота:
Наследуется по аутосомно-рецессивному типу (болеют дети здоровых родителей).
Болезнь

Больные испытывают затруднение при вставании с пола, помогая себе при этом
Больные испытывают затруднение при вставании с пола, помогая себе при этом
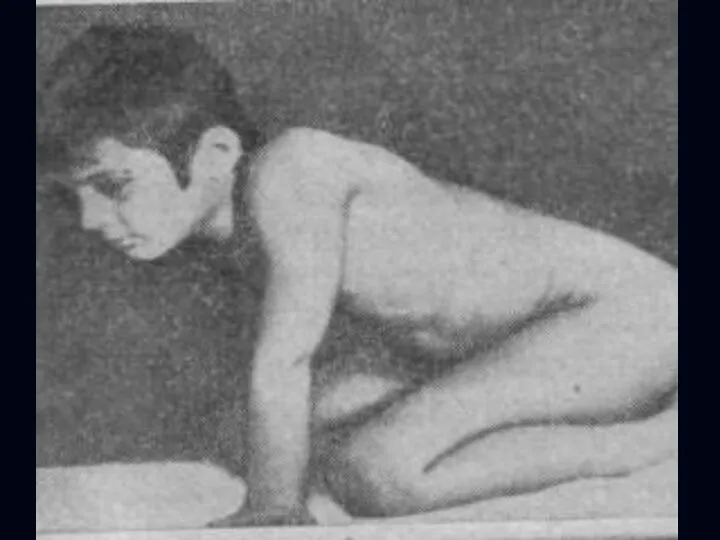

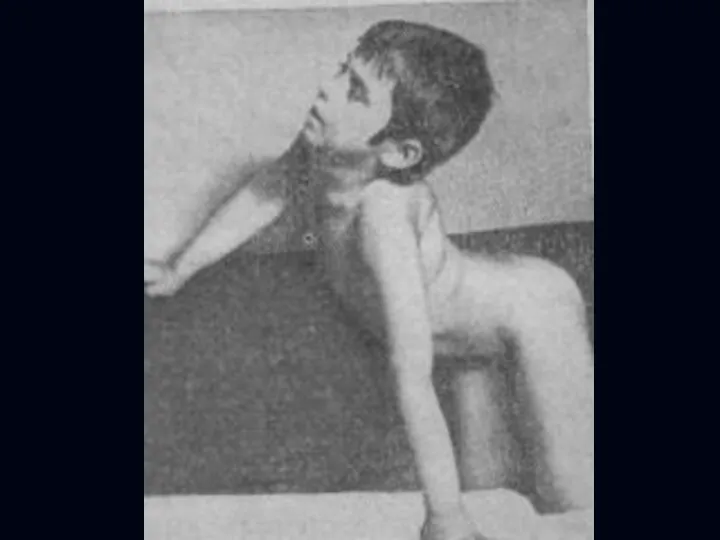

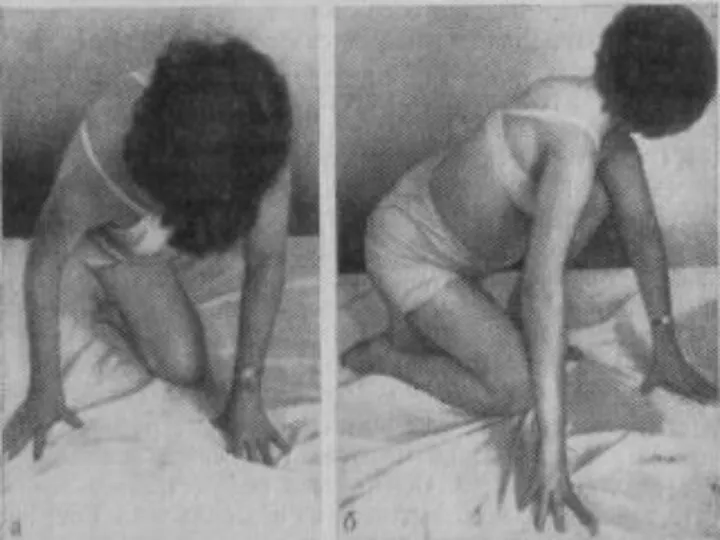
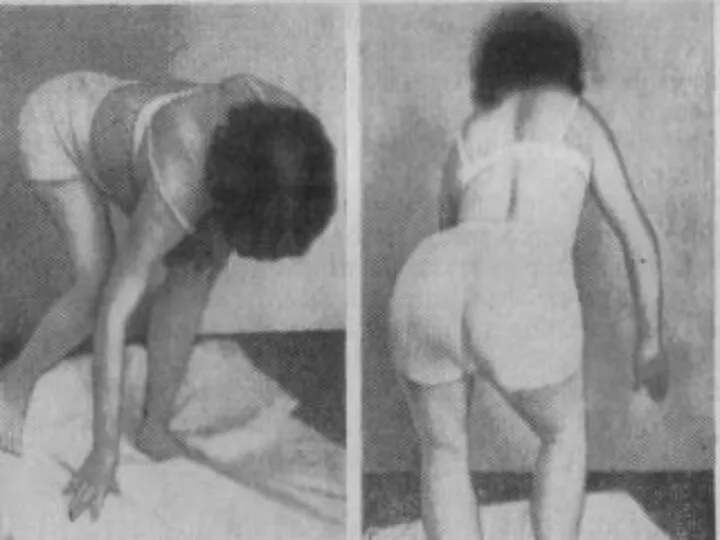

Лице-лопаточно-плечевая форма Ландузи-Дежерина.
Начало заболевания с 7 - 15 лет.
Слабость и атрофии
Лице-лопаточно-плечевая форма Ландузи-Дежерина.
Начало заболевания с 7 - 15 лет.
Слабость и атрофии

Симптом «крыловидных лопаток», горизонтальное расположение ключиц.
Характерны толстые, выпяченные вперед губы («губы
Симптом «крыловидных лопаток», горизонтальное расположение ключиц.
Характерны толстые, выпяченные вперед губы («губы
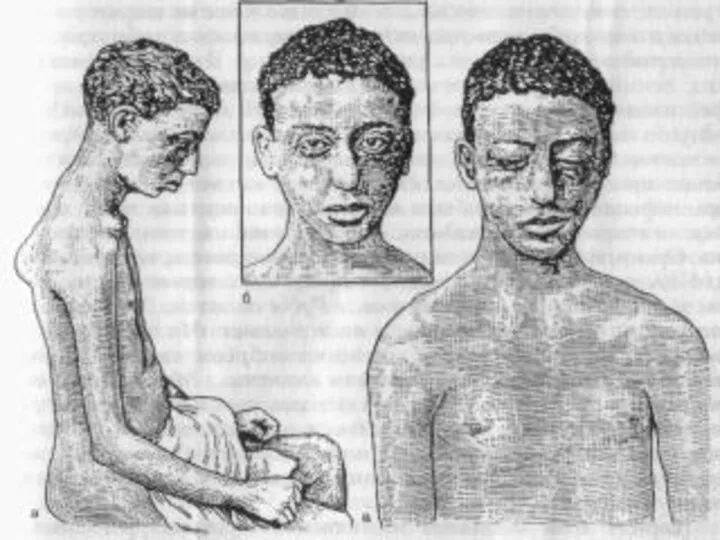

Спинальная амиотрофия Вердига – Гоффмана.
Выделяют формы:
Врожденная форма.
Ранняя детская форма.
Поздняя форма.
Спинальная амиотрофия Вердига – Гоффмана.
Выделяют формы:
Врожденная форма.
Ранняя детская форма.
Поздняя форма.

Врожденная форма:
Начало внутриутробно.
Слабое шевеление плода.
При рождении вялый паралич в проксимальных
Врожденная форма:
Начало внутриутробно.
Слабое шевеление плода.
При рождении вялый паралич в проксимальных

Ранняя детская форма:
Начало до 1,5 лет.
После интеркурретного заболевания ребенок начинает терять
Ранняя детская форма:
Начало до 1,5 лет.
После интеркурретного заболевания ребенок начинает терять

Поздняя форма:
Начало в 1,5 – 2 года.
Вялые параличи проксимальных отделов ног,
Поздняя форма:
Начало в 1,5 – 2 года.
Вялые параличи проксимальных отделов ног,
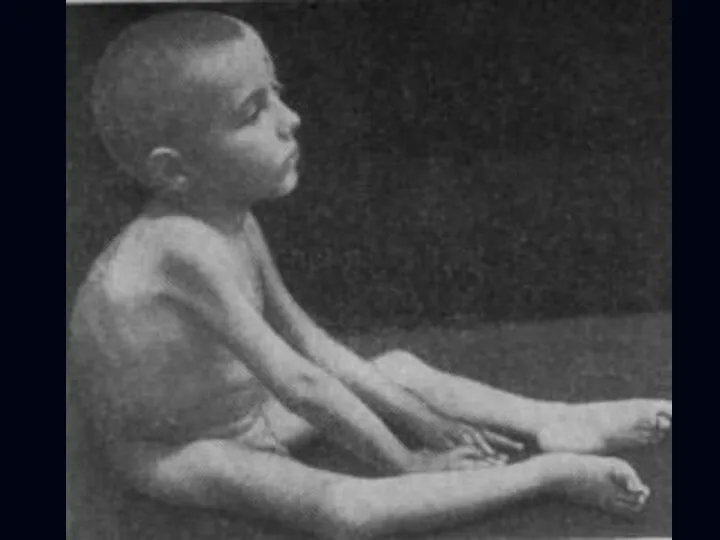
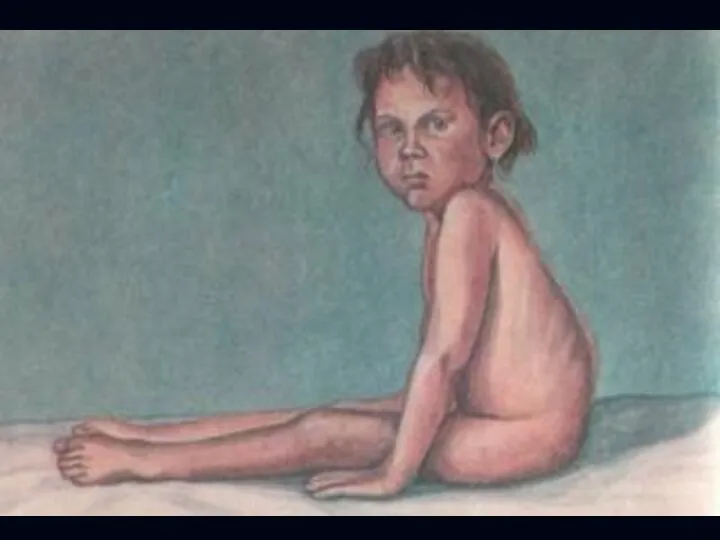
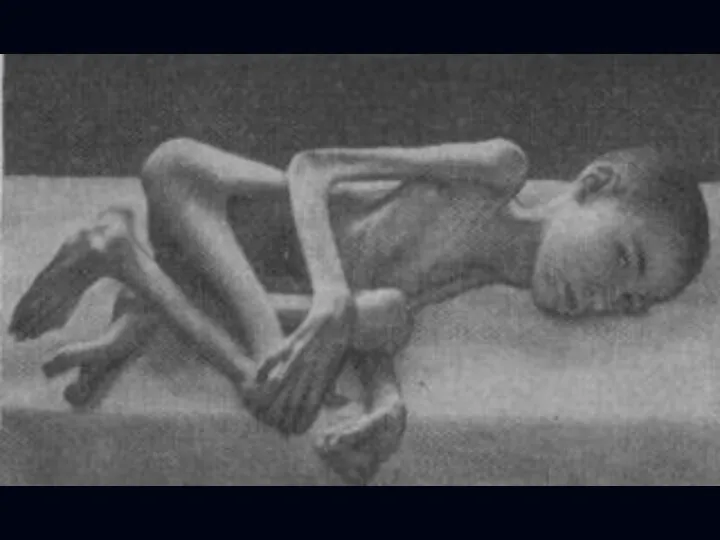
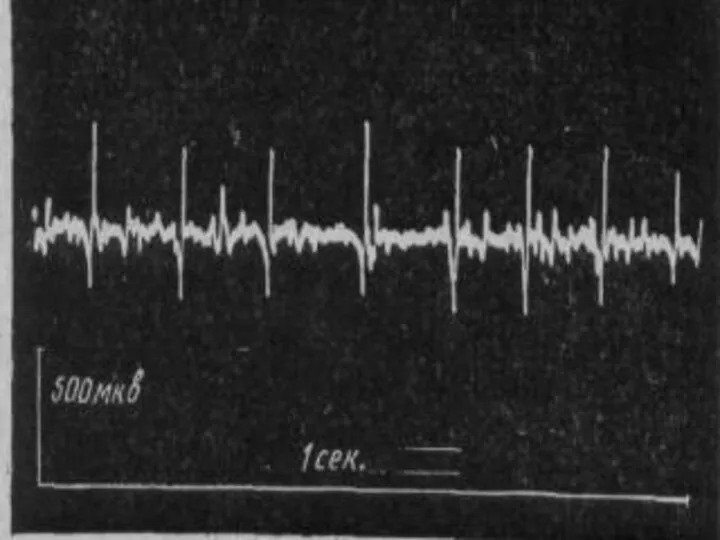

Спинальная амиотрофия Кугельберга-Веландера
(псевдомиопатическая).
Тип наследования аутосомно-рецессивный.
Начало в 6 лет.
Течение доброкачественное, ещё в
Спинальная амиотрофия Кугельберга-Веландера
(псевдомиопатическая).
Тип наследования аутосомно-рецессивный.
Начало в 6 лет.
Течение доброкачественное, ещё в

Невральная амиотрофия Шарко-Мари
Тип наследования аутосомно-доминантный.
Начальные проявления с 13-17 лет.
Характерны дистальные парезы,
Невральная амиотрофия Шарко-Мари
Тип наследования аутосомно-доминантный.
Начальные проявления с 13-17 лет.
Характерны дистальные парезы,

Атрофия мышц голеней и нижней трети бедер.
Ноги по виду напоминают «ноги
Атрофия мышц голеней и нижней трети бедер.
Ноги по виду напоминают «ноги
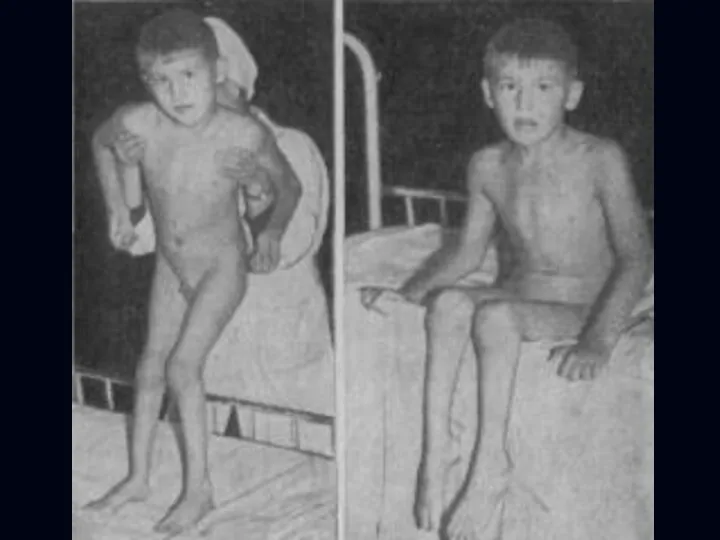

Миастения:
Появляется утомляемость, распространяется на мышцы: губ, жевательные, глазодвигательные, глотательные.
Наблюдается птоз, диплопия,
Миастения:
Появляется утомляемость, распространяется на мышцы: губ, жевательные, глазодвигательные, глотательные.
Наблюдается птоз, диплопия,

Утомляемость появляется в мышцах конечностей.
Наблюдается увеличение вилочковой железы.
Заболевание имеет тенденцию к
Наблюдается увеличение вилочковой железы.
Заболевание имеет тенденцию к

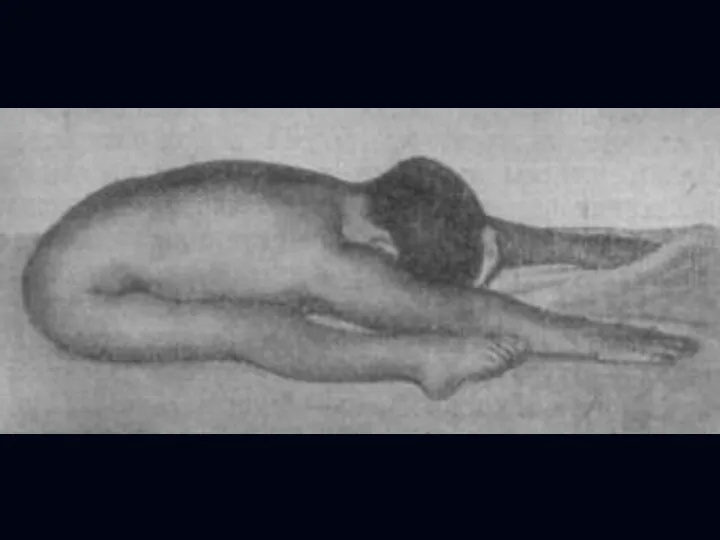
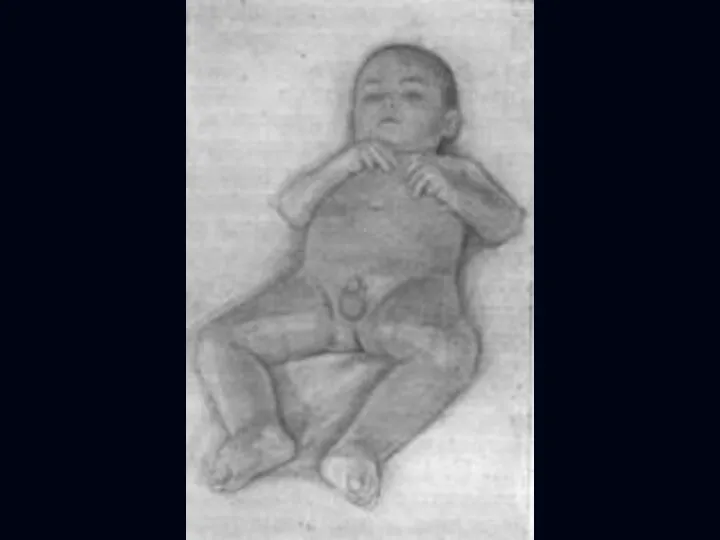
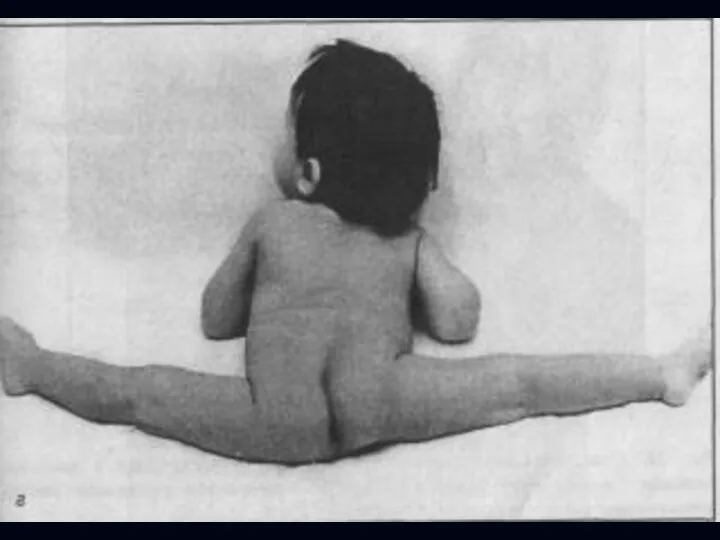
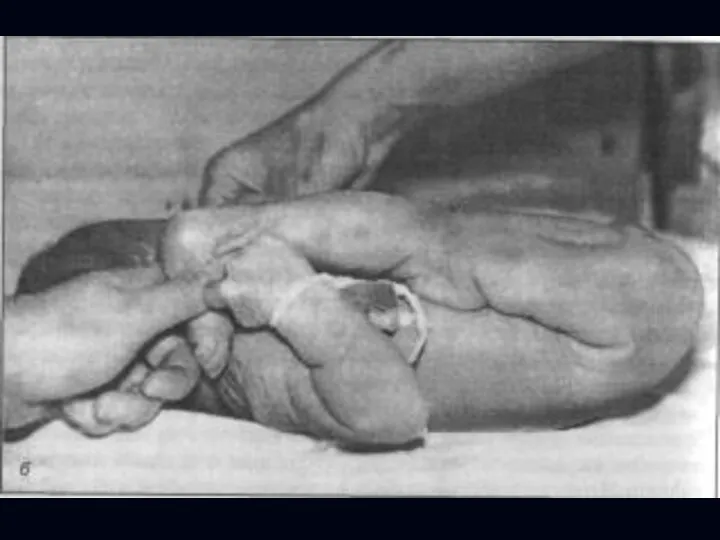
Врожденная миатония Оппенгейма:
С рождения – мало активных движений.
Резкая гипотония или полная
Врожденная миатония Оппенгейма:
С рождения – мало активных движений.
Резкая гипотония или полная

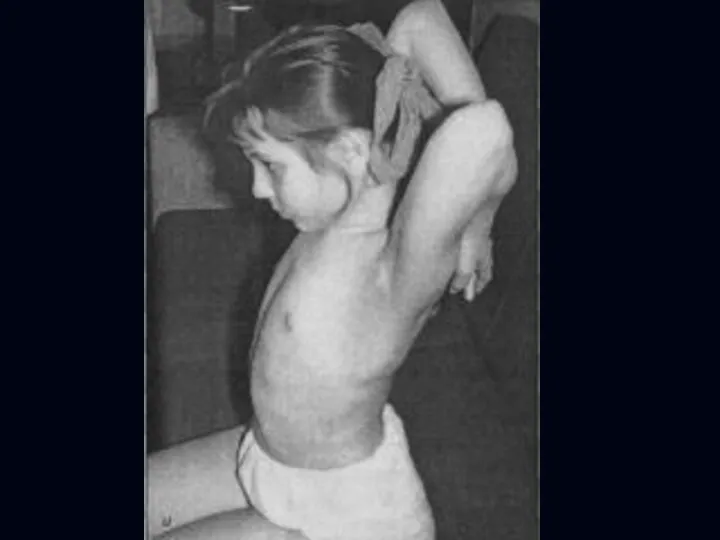
Отсутствуют также гипертрофия и псевдогипертрофия.
Лицевые мышцы поражаются редко.
Психика сохранена.
Тазовых и сенсорных
Отсутствуют также гипертрофия и псевдогипертрофия.
Лицевые мышцы поражаются редко.
Психика сохранена.
Тазовых и сенсорных


Пароксизмальная миоплегия
- генетически детерминированные нервно-мышечные заболевания, обусловленные нарушениями обмена
Пароксизмальная миоплегия
- генетически детерминированные нервно-мышечные заболевания, обусловленные нарушениями обмена

Гипокалиемическая форма:
1. Начало от 10 до 16 лет.
2. Возникновение приступов
Гипокалиемическая форма:
1. Начало от 10 до 16 лет.
2. Возникновение приступов

6. При парциальном приступе мышечная слабость и парез или плегия развивается
6. При парциальном приступе мышечная слабость и парез или плегия развивается

Гиперкалиемическая форма:
1. Начало от 10 до 18 лет.
2. Возникают приступы пароксизмаль-ного
Гиперкалиемическая форма:
1. Начало от 10 до 18 лет.
2. Возникают приступы пароксизмаль-ного

5. В последующем развивается мышечная слабость, гипотония мышц, снижение сухожильных рефлексов.
6.
5. В последующем развивается мышечная слабость, гипотония мышц, снижение сухожильных рефлексов.
6.

Нормокалиемическая форма:
1. Начало в первые 10 лет жизни.
2. Сочетание признаков
Нормокалиемическая форма:
1. Начало в первые 10 лет жизни.
2. Сочетание признаков

Дополнительные методы обследования:
Электромиография (ЭМГ).
Биопсия скелетной мышцы.
Биохимический анализ крови (БАК).
Компьютерная томография скелетных
Дополнительные методы обследования:
Электромиография (ЭМГ).
Биопсия скелетной мышцы.
Биохимический анализ крови (БАК).
Компьютерная томография скелетных

Лечение прогрессирующих мышечных миопатий:
Для улучшения трофики: АТФ.
Антихолинэстеразные препараты: галантамин.
Витаминотерапия:
витамины
Лечение прогрессирующих мышечных миопатий:
Для улучшения трофики: АТФ.
Антихолинэстеразные препараты: галантамин.
Витаминотерапия:
витамины

Лечение прогрессирующих мышечных амиотрофий:
Для улучшения трофики: АТФ, глютаминовая кислота, метионин, лейцин.
Антихолинэстеразные
Лечение прогрессирующих мышечных амиотрофий:
Для улучшения трофики: АТФ, глютаминовая кислота, метионин, лейцин.
Антихолинэстеразные

Лечение невральной амиотрофии Шарко-Мари:
Антихолинэстеразные препараты: прозерин, дибазол.
Для улучшения трофики: АТФ.
ЛФК,
Лечение невральной амиотрофии Шарко-Мари:
Антихолинэстеразные препараты: прозерин, дибазол.
Для улучшения трофики: АТФ.
ЛФК,

Лечение миастении:
Антихолинэстеразные препараты:
прозерин, местинон, оксазил.
Препарат, увеличивающий нервно-мышечную передачу – хлористый
Лечение миастении:
Антихолинэстеразные препараты:
прозерин, местинон, оксазил.
Препарат, увеличивающий нервно-мышечную передачу – хлористый

Лечение пароксизмальной миоплегии:
Во время приступа.
Высокие дозы хлорида калия внутрь (10-15 г
Лечение пароксизмальной миоплегии:
Во время приступа.
Высокие дозы хлорида калия внутрь (10-15 г

В межприступный период:
1) Диета с высоким содержанием калия и ограничением углеводов
В межприступный период:
1) Диета с высоким содержанием калия и ограничением углеводов

Литература:
1. «Детская неврология», Л.О. Бадалян, 2003г.
2. «Наследственные болезни нервной системы» Ю.Е.
Литература:
1. «Детская неврология», Л.О. Бадалян, 2003г.
2. «Наследственные болезни нервной системы» Ю.Е.

4. «Неврология детского возраста», А.С. Петрухин, 2004г.
5. «Нервные болезни», С.И. Гусев,
4. «Неврология детского возраста», А.С. Петрухин, 2004г.
5. «Нервные болезни», С.И. Гусев,
 Полное построение алгоритма. Часть 1. Задача коммивояжера
Полное построение алгоритма. Часть 1. Задача коммивояжера Домашников Борис Федорович 1924 - 2003
Домашников Борис Федорович 1924 - 2003 Электропитание устройств и систем телекоммуникации. Инверторы
Электропитание устройств и систем телекоммуникации. Инверторы Боги Древнего Египта
Боги Древнего Египта Принтеры
Принтеры  ВИЧ-инфекция. Парэнтеральные гепатиты
ВИЧ-инфекция. Парэнтеральные гепатиты Trade
Trade  Особенности поражения АОХВ с преимущественного цитотоксическим действием
Особенности поражения АОХВ с преимущественного цитотоксическим действием Орифлэйм - построй жизнь, которой ты будешь гордиться
Орифлэйм - построй жизнь, которой ты будешь гордиться Пятиклассник. Первые шаги. Школы начальной ты выпускник! В тайны наук самых первых проник. Много трудов у тебя позади - Больше их б
Пятиклассник. Первые шаги. Школы начальной ты выпускник! В тайны наук самых первых проник. Много трудов у тебя позади - Больше их б Общаться с ребенком. Как?
Общаться с ребенком. Как? Состояние вещества, типы связи
Состояние вещества, типы связи  Электротехника в школе
Электротехника в школе Декоративно-прикладное творчество. Хохломская роспись
Декоративно-прикладное творчество. Хохломская роспись СЛОВО 9 КЛ
СЛОВО 9 КЛ Правовое регулирование инновационной деятельности в РК
Правовое регулирование инновационной деятельности в РК Строки. Пример использования С и С++
Строки. Пример использования С и С++ Физическое воспитание
Физическое воспитание День конституции РФ
День конституции РФ Презентация "Энергетика одна из важнейших отраслей экономики" - скачать презентации по Экономике
Презентация "Энергетика одна из важнейших отраслей экономики" - скачать презентации по Экономике ТОКСИКОЛОГИЯ
ТОКСИКОЛОГИЯ Рождество и введение во храм Пресвятой Богородицы
Рождество и введение во храм Пресвятой Богородицы ОСНОВНЫЕ КОНЦЕПЦИИ СОВРЕМЕННОЙ НАУКИ Подготовили: Доценко Юлия Тс01/1301 Опутина Анна Тс01/1301
ОСНОВНЫЕ КОНЦЕПЦИИ СОВРЕМЕННОЙ НАУКИ Подготовили: Доценко Юлия Тс01/1301 Опутина Анна Тс01/1301 БАЗЫ ДАННЫХ. ACCESS 2007 Запросы
БАЗЫ ДАННЫХ. ACCESS 2007 Запросы Этапы развития квантовой механики.
Этапы развития квантовой механики. Глава 2. Экономика домохозяйства 12. Рынок труда
Глава 2. Экономика домохозяйства 12. Рынок труда Создай свою Игру и получи поток ВИП-Клиентов
Создай свою Игру и получи поток ВИП-Клиентов Русская культура VI-XIII
Русская культура VI-XIII