Выполнила: студентка 4 гр. 3 мед. факультета Сыченко Д.Е. Преподаватель: Гречанина Ю.Б Синдром Марфана
-
Выполнила: студентка 4 гр. 3 мед. факультета Сыченко Д.Е. Преподаватель: Гречанина Ю.Б Синдром Марфана

Содержание
- 2. Синдром Марфана (СМ), или Марфана-Ашара – это наследственное заболевание соединительной ткани с преимущественным поражением сердечно-сосудистой системы,
- 3. Существует интересный факт, что первая девушка модель - Лесли Хорнби, которая послужила прототипом образа всех моделей,
- 4. Н. Паганини, Ш. де Голль, Г.Х. Андерсен, А. Линкольн.
- 5. Этиология и патогенез. СМ относят к наследственным болезням соединительной ткани с аутосомно-доминантным типом наследования. Молекулярной основой
- 6. При вступлении в брак 1 больного и 1 здорового родителя вероятность рождения больных детей – 50%.
- 7. В настоящее время в различных семьях идентифицировано более 550 мутаций. Среди обнаруженных мутаций в гене FBN1
- 8. Классификация. I. Форма: 1. Стертая: слабо выраженные изменения в одной, двух системах. 2. Выраженная: а) слабо
- 9. IV. Клинические варианты: 1. Болезнь Марфана (присутствие трех классических признаков, семейный характер заболевания). 2. Синдром Марфана
- 10. Клиника. Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ многосистемна и разнообразна. При этом
- 11. Наиболее частая сердечная патология при СМ – недостаточность митрального клапана. Обычно наблюдается поражение эластических структур створок
- 12. Патологические процессы со стороны аорты при СМ. Расширяется корень аорты, ее клапанное кольцо и синус Вальсальвы.
- 13. Проявления со стороны скелета наблюдаются у 2/3 пациентов. высокий рост, астеническое телосложение, долихостеномелию, долихоцефалию, прогнатию, "готическое"
- 16. Известны следующие фенотипические диагностические тесты СМ: - соотношение кисть-рост > 11%; - отношение размаха рук к
- 17. Весьма часто при СМ бывают положительными тесты на арахнодактилию: а) тест большого пальца Steinberg: согнутый 1-й
- 19. Офтальмологиеские признаки. Наиболее часто встречается миопия различной степени, гипоплазия радужки, цилиарной мышцы и пигментной каймы зрачкового
- 20. Нередко при СМ наблюдаются поражения со стороны других органов и систем: Легких: поликистоз, эмфизема, спонтанный пневмоторакс);
- 21. Диагностика. Дополнительные методы исследования. 1. Лабораторные. Наиболее точным лабораторным признаком СМ является генетическая идентификация мутаций в
- 22. Диагностические признаки синдрома Марфана.
- 24. Согласно «Гентской нозологии» для диагноза СМ необходимо, как минимум, наличие по одному большому критерию в двух
- 25. Лечение. Консервативное. Так как ведущая причина смерти больных СМ - разрыв расслаивающей аневризмы аорты, то консервативное
- 26. Хирургическое. В настоящее время при СМ в основном применяется два типа вмешательств на аорте: комбинированная трансплантация
- 27. Синдром Марфана и беременность. Беременность при СМ опасна, по крайней мере, по двум причинам. 1. Имеется
- 28. Диспансерное наблюдение. В целях предотвращения прогрессирования заболевания и профилактики осложнений необходимо: 1. Регулярное наблюдение квалифицированных специалистов
- 29. Прогноз. Продолжительность и качество жизни больных СМ в основном зависит от объема и выраженности поражения сердечно-сосудистой
- 31. Скачать презентацию

Синдром Марфана (СМ), или Марфана-Ашара – это наследственное заболевание соединительной ткани
Синдром Марфана (СМ), или Марфана-Ашара – это наследственное заболевание соединительной ткани

Существует интересный факт, что первая девушка модель - Лесли Хорнби, которая
Существует интересный факт, что первая девушка модель - Лесли Хорнби, которая

Н. Паганини, Ш. де Голль, Г.Х. Андерсен,
А. Линкольн.
Н. Паганини, Ш. де Голль, Г.Х. Андерсен,
А. Линкольн.

Этиология и патогенез.
СМ относят к наследственным болезням соединительной ткани с аутосомно-доминантным
Этиология и патогенез.
СМ относят к наследственным болезням соединительной ткани с аутосомно-доминантным
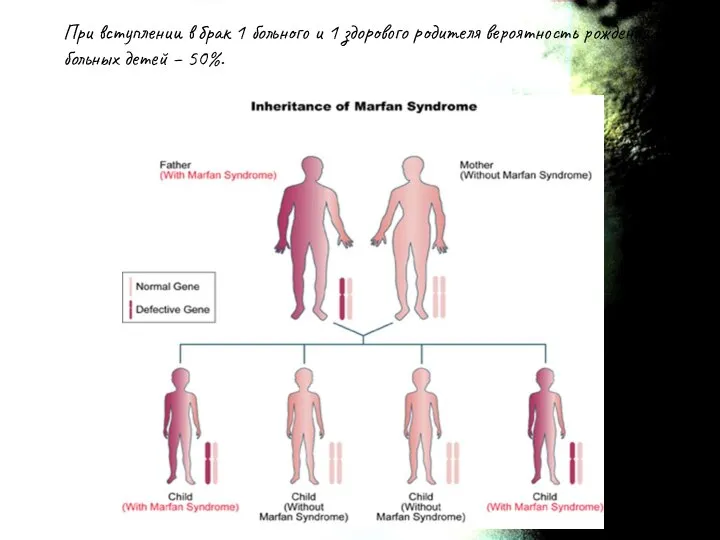
При вступлении в брак 1 больного и 1 здорового родителя вероятность
При вступлении в брак 1 больного и 1 здорового родителя вероятность

В настоящее время в различных семьях идентифицировано более 550 мутаций.
Среди обнаруженных
В настоящее время в различных семьях идентифицировано более 550 мутаций.
Среди обнаруженных

Классификация.
I. Форма:
1. Стертая: слабо выраженные изменения в одной, двух системах.
2. Выраженная:
Классификация.
I. Форма:
1. Стертая: слабо выраженные изменения в одной, двух системах.
2. Выраженная:

IV. Клинические варианты:
1. Болезнь Марфана (присутствие трех классических признаков, семейный характер
IV. Клинические варианты:
1. Болезнь Марфана (присутствие трех классических признаков, семейный характер

Клиника.
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ многосистемна
Клиника.
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ многосистемна

Наиболее частая сердечная патология при СМ –
недостаточность митрального клапана. Обычно наблюдается
Наиболее частая сердечная патология при СМ –
недостаточность митрального клапана. Обычно наблюдается

Патологические процессы со стороны аорты при СМ. Расширяется корень аорты, ее
Патологические процессы со стороны аорты при СМ. Расширяется корень аорты, ее

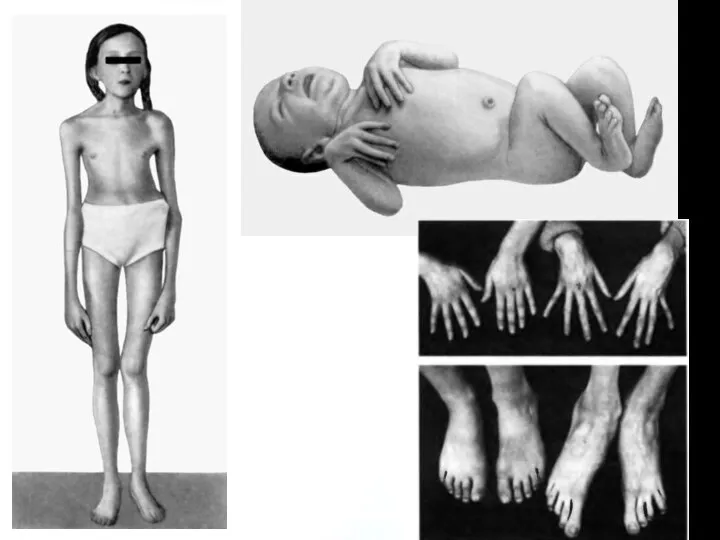
Проявления со стороны скелета наблюдаются у 2/3 пациентов.
высокий рост, астеническое
Проявления со стороны скелета наблюдаются у 2/3 пациентов.
высокий рост, астеническое
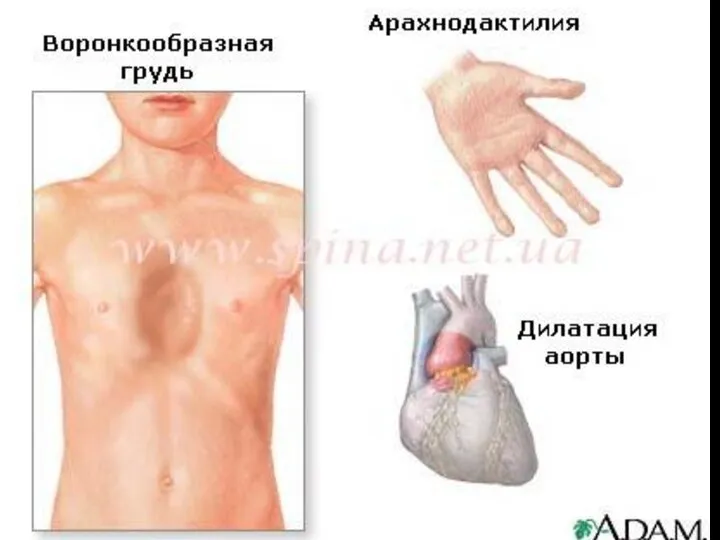

Известны следующие фенотипические диагностические тесты СМ:
- соотношение кисть-рост > 11%;
- отношение
Известны следующие фенотипические диагностические тесты СМ:
- соотношение кисть-рост > 11%;
- отношение

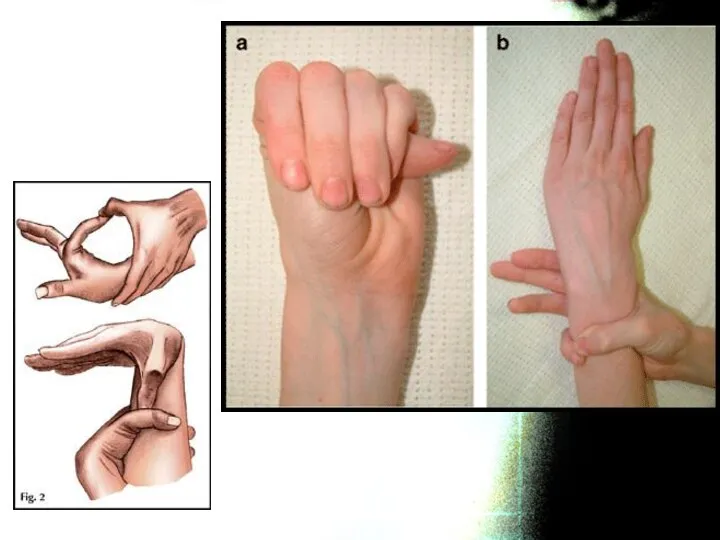
Весьма часто при СМ бывают положительными тесты на арахнодактилию:
а) тест большого
Весьма часто при СМ бывают положительными тесты на арахнодактилию:
а) тест большого

Офтальмологиеские признаки.
Наиболее часто встречается миопия различной степени, гипоплазия радужки, цилиарной мышцы
Офтальмологиеские признаки.
Наиболее часто встречается миопия различной степени, гипоплазия радужки, цилиарной мышцы

Нередко при СМ наблюдаются поражения со стороны других органов и систем:
Нередко при СМ наблюдаются поражения со стороны других органов и систем:

Диагностика. Дополнительные методы исследования.
1. Лабораторные.
Наиболее точным лабораторным признаком СМ является генетическая
Диагностика. Дополнительные методы исследования.
1. Лабораторные.
Наиболее точным лабораторным признаком СМ является генетическая

Диагностические признаки синдрома Марфана.
Диагностические признаки синдрома Марфана.
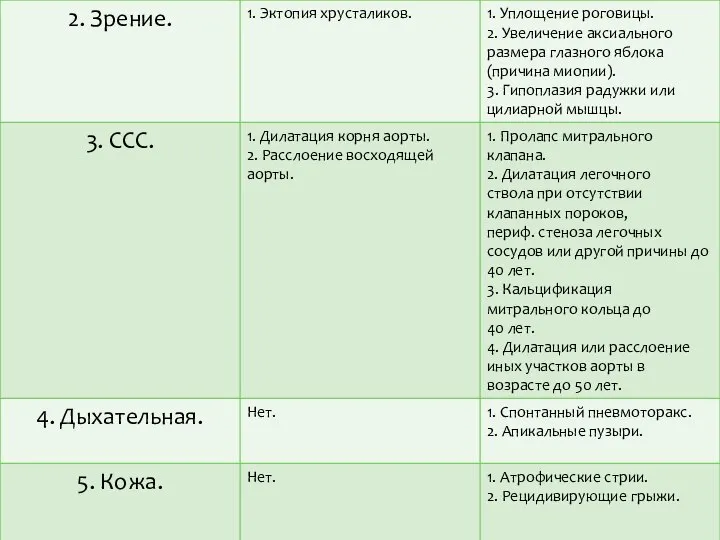

Согласно «Гентской нозологии» для диагноза СМ необходимо, как минимум,
наличие по одному
Согласно «Гентской нозологии» для диагноза СМ необходимо, как минимум,
наличие по одному

Лечение.
Консервативное.
Так как ведущая причина смерти больных СМ - разрыв расслаивающей
Лечение.
Консервативное.
Так как ведущая причина смерти больных СМ - разрыв расслаивающей

Хирургическое.
В настоящее время при СМ в основном применяется два типа
Хирургическое.
В настоящее время при СМ в основном применяется два типа

Синдром Марфана и беременность.
Беременность при СМ опасна, по крайней мере, по
Синдром Марфана и беременность.
Беременность при СМ опасна, по крайней мере, по

Диспансерное наблюдение.
В целях предотвращения прогрессирования заболевания и профилактики осложнений необходимо:
1. Регулярное
Диспансерное наблюдение.
В целях предотвращения прогрессирования заболевания и профилактики осложнений необходимо:
1. Регулярное

Прогноз.
Продолжительность и качество жизни больных СМ в основном зависит от объема
Прогноз.
Продолжительность и качество жизни больных СМ в основном зависит от объема
 Знаете ли вы простое и сложное предложение? - презентация для начальной школы_
Знаете ли вы простое и сложное предложение? - презентация для начальной школы_ Презентация Базовые модели систем
Презентация Базовые модели систем Аналіз діяльності показників роботи Головного управління Держгеокадастру у Закарпатській області
Аналіз діяльності показників роботи Головного управління Держгеокадастру у Закарпатській області Как появился мобильный телефон
Как появился мобильный телефон Технические аспекты использования интернета
Технические аспекты использования интернета Подготовка к ГИА по обществознанию « Сфера духовной культуры»
Подготовка к ГИА по обществознанию « Сфера духовной культуры» Религия и искусство в системе культуры
Религия и искусство в системе культуры L’image des français dans le monde
L’image des français dans le monde Natsionalnaya_kukhnya
Natsionalnaya_kukhnya Brake system
Brake system Военная экономика России: смена парадигмы? В.Б. Зацепин к.воен.н., с.н.с
Военная экономика России: смена парадигмы? В.Б. Зацепин к.воен.н., с.н.с КООРДИНАЦИЯ
КООРДИНАЦИЯ  Предложение по организации городских пространств в рамках проекта «Формирование комфортной городской среды» для г. Тарко-Сале
Предложение по организации городских пространств в рамках проекта «Формирование комфортной городской среды» для г. Тарко-Сале Итоги работы 5 класса - презентация для начальной школы
Итоги работы 5 класса - презентация для начальной школы Презентация Маркетинговые исследования
Презентация Маркетинговые исследования Становление и развитие органов внутренних дел в 1917-1924гг
Становление и развитие органов внутренних дел в 1917-1924гг Прямой поперечный изгиб
Прямой поперечный изгиб Методологические подходы к управлению Выполнили студенты группы Т-1210 Тагиев Р Ишонов Ф
Методологические подходы к управлению Выполнили студенты группы Т-1210 Тагиев Р Ишонов Ф Избирательное право в РФ
Избирательное право в РФ На юге Европы 3 КЛАСС - презентация для начальной школы_
На юге Европы 3 КЛАСС - презентация для начальной школы_ Педагогические технологии Педагогическая технология - это такое построение деятельности педагога, в котором все действия находя
Педагогические технологии Педагогическая технология - это такое построение деятельности педагога, в котором все действия находя Звук и буквы и й - презентация для начальной школы_
Звук и буквы и й - презентация для начальной школы_ РОЛЬ МОРАЛЬНО-ПСИХОЛОГИЧЕСКОГО ФАКТОРА В УСЛОВИЯХ СОВРЕМЕННОЙ ВОЙНЫ
РОЛЬ МОРАЛЬНО-ПСИХОЛОГИЧЕСКОГО ФАКТОРА В УСЛОВИЯХ СОВРЕМЕННОЙ ВОЙНЫ Экономическое районирование России Группа Э122б Восканян Светлана,Багатурия Гульнаяра
Экономическое районирование России Группа Э122б Восканян Светлана,Багатурия Гульнаяра Государство в политической системе
Государство в политической системе Энергосбережение в зданиях. Объемно-планировочные решения
Энергосбережение в зданиях. Объемно-планировочные решения Игра. Застольный этикет (2)
Игра. Застольный этикет (2) Зимние виды спорта
Зимние виды спорта