- Болезни, вызванные мутацией отдельного гена (менделевские)

Содержание
- 2. Моногенные болезни
- 3. Структура и экспрессия гена I II III Интроны II III I мРНК Белок Сплайсинг I II
- 4. Типы мутаций Делеция Инсерция Дупликация Инверсия Миссенс Нонсенс Со сдвигом рамки считывания Сплайсинг Экспансия тринкулеотидных повторов
- 5. Патогенетические типы мутаций Мутации, ведущие к потере функции (ингибирование процессов транскрипции, трансляции, нарушение структуры и функции
- 6. http://www.ncbi.nlm.nih.gov/Omim/
- 7. Каждому фенотипу в OMIM присвоен уникальный номер из шести цифр, первая цифра которого указывает способ наследования
- 8. Символ (*) перед номером указывает гены с известной последовательностью. Символ (#) перед номером указывает, что в
- 9. OMIM Statistics for October 8, 2010 Number of Entries
- 10. OMIM Entry Statistics
- 11. OMIM Entry Statistics
- 12. Для развития заболевания достаточно получить один мутантный аллель От одного из родителей или вследствие мутации de
- 13. Для развития заболевания индивидуумы должны получить два мутантных аллеля – по одному от каждого из родителей.
- 14. Частный случай рецессивного наследования, болеют мужчины получившие с Х хромосомой мутантный аллель. Женщины не болеют, так
- 15. Дислипопротедемии Первичная Вторичная (на фоне соматических заболеваний) Наследственная Обусловленная факторами внешней среды (питание) Моногенная Полигенная
- 16. Моногенные болезни, с нарушениями липидного обмена Болезни накопления липидов (тезаурисмозы) - внутриклеточные липидозы, при которых наблюдается
- 17. Семейная гиперхолестеринемия (143890) Повышение уровня ЛПНП в крови Частота гетерозиготной формы в популяции 1:500 Среди лиц
- 18. ВНУТРИКЛЕТОЧНЫЙ ЦИКЛ РЕЦЕПТОРА ЛПНП (по J.L. Goldstein, M.S. Brown, J. Cell. Sci., Suppl. N 3, 1985
- 19. Рецептор ЛПНП синтезируется в виде предшественника с массой 120 кДа на рибосомах, связанных с эндоплазматическим ретикулюмом
- 20. Структура рецептора ЛПНП Наиболее надежным методом дифференциальной диагностики СГ является определение мутаций в гене рецептора ЛПНП,
- 21. ПОЛИМОРФИЗМЫ КОДИРУЮЩЕЙ ОБЛАСТИ ГЕНА РЕЦЕПТОРА ЛНП В ПОПУЛЯЦИИ САНКТ-ПЕТЕРБУРГА Полиморфизмы - варианты нуклеотидной последовательности, не ведущие
- 22. Семейная гиперхиломикронемия Аккумуляция ХМ в плазме крови может быть обусловлена двумя основными причинами: — генетическим отсутствием
- 23. ГИПЕРЛИПОПРОТЕИНЕМИЯ ТИП 1 (+238600) Синонимы LIPOPROTEIN LIPASE DEFICIENCY LPL DEFICIENCY HYPERCHYLOMICRONEMIA, FAMILIAL HYPERLIPEMIA, IDIOPATHIC, BURGER-GRUTZ TYPE
- 24. АПОЛИПОПРОТЕИН C-II ДЕФИЦИТ (207750) Синонимы HYPERLIPOPROTEINEMIA, TYPE IB C-II ANAPOLIPOPROTEINEMIA APOC2 DEFICIENCY Ген локализован 19q13.2, состоит
- 25. TANGIER DISEASE (205400); TGD Синонимы HIGH DENSITY LIPOPROTEIN DEFICIENCY, TYPE 1; HDLDT1 HIGH DENSITY LIPOPROTEIN DEFICIENCY,
- 26. Абеталипопротеинемия (200100) Генный локус 4q22-q24 Нарушение всасывания и транспорта жиров Недостаточность или отсутствие бета-липопротеинов Недостаточность высших
- 27. Ожирение С-м Карпентера (201000) С-м Альстрёма (203800) С-м Барде-Бидля (209900) С-м Бьерсона-Форсмана-Лемана (301900), Х-сцепленный рецессивный Карликовость
- 28. Липоатрофия, снижение или отсутствие подкожного жирового слоя Синдром Вернера (277700) Синдром Клиппеля-Треноне-Вебера (176670) Синдром Коккейна (216400)
- 29. Прогерия, синдром Гетчинсона-Гилфорда Низкий рост и вес Отсутствие подкожного жирового слоя Тотальная алопеция Маленькое лицо Микрогнатия
- 30. Синдром Вернера, прогерия взрослых Начало заболевания в 15-30 лет Ювенильная катаракта Преждевременное поседение и облысение Склеродермия
- 31. Липоматоз Наследование аутосомно-доминантное. Фенотипически проявляется безболезненными медленно растущими липомами. Необходимо дифференцировать с синдромом Маделунга (множественный симметричный
- 32. Себоцистоматоз причиной развития себоцистоматоза являются мутации в гене кератина 17 (KRT17, MIM 148069), которые также обнаружены
- 33. Генотерапия Лечение СГХС. Резекция части печени, введение в клетки гена. Возврат клеток в организм. Исследователи из
- 34. Вульгарный ихтиоз
- 35. Х-сцепленный рецессивный ихтиоз
- 36. Классификация наследственных заболеваний обмена веществ нарушения обмена аминокислот (фенилкетонурия и др.); нарушения обмена углеводов (гликогеновая болезнь,
- 37. Признаки, требующие исключения НБО В неонатальном периоде и на первом году жизни Рвота, дегидротация, желтуха ,
- 38. После первого года жизни Умственная отсталость неясной этиологии. УО с задержкой физического развития, судорожными припадками, интоксикацией,
- 39. Симптомы и признаки болезней обмена веществ у детей Центральная нервная система Летаргия, вялое сосание Повышенная возбудимость
- 40. Органы дыхания и кровообращения Апноэ Тахипноэ Респираторный дистресс Нарушение функции печени Желтуха Гипокоагуляция Гепатомегалия Прочие нарушения
- 41. Врожденные дефекты метаболизма А. Острое начало в неонатальный период Заболевания встречаются редко, но все вместе значительно
- 42. Б. Скрытое начало в период младенчества или детства Некоторые заболевания неизлечимы и являются фатальными Большинство из
- 43. Болезни обмена веществ у новорожденных НАРУШЕНИЕ ОБМЕНА УГЛЕВОДОВ Галактоземия Дефицит фруктозо-1,6-дифосфатазы Гликогенозы (1А и В типы)
- 44. НАРУШЕНИЯ ОБМЕНА ПИРУВАТА И ТРАНСПОРТА ЭЛЕКТРОНОВ В ДЫХАТЕЛЬНОЙ ЦЕПИ Дефицит пируваткарбоксилазы Дефицит пируватдегидрогеназы Дефицит дыхательных цепей
- 45. Заболевания, на которые проводится неонатальный скрининг в России Фенилкетонурия Врождённый гипотиреоз Муковисцидоз Галактоземия Адреногенитальный синдром
- 47. Скачать презентацию

Моногенные болезни
Моногенные болезни
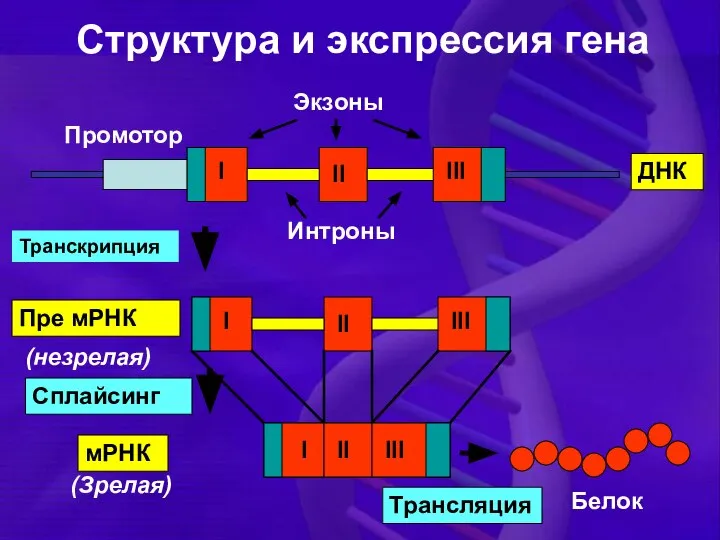
Структура и экспрессия гена
I
II
III
Интроны
II
III
I
мРНК
Белок
Сплайсинг
I
II
III
Промотор
Пре мРНК
ДНК
Транскрипция
(незрелая)
(Зрелая)
Экзоны
Трансляция
Структура и экспрессия гена
I
II
III
Интроны
II
III
I
мРНК
Белок
Сплайсинг
I
II
III
Промотор
Пре мРНК
ДНК
Транскрипция
(незрелая)
(Зрелая)
Экзоны
Трансляция

Типы мутаций
Делеция
Инсерция
Дупликация
Инверсия
Миссенс
Нонсенс
Со сдвигом рамки считывания
Сплайсинг
Экспансия тринкулеотидных повторов
Типы мутаций
Делеция
Инсерция
Дупликация
Инверсия
Миссенс
Нонсенс
Со сдвигом рамки считывания
Сплайсинг
Экспансия тринкулеотидных повторов

Патогенетические типы
мутаций
Мутации, ведущие к потере функции (ингибирование процессов транскрипции, трансляции, нарушение
Патогенетические типы
мутаций
Мутации, ведущие к потере функции (ингибирование процессов транскрипции, трансляции, нарушение
http://www.ncbi.nlm.nih.gov/Omim/
http://www.ncbi.nlm.nih.gov/Omim/

Каждому фенотипу в OMIM присвоен уникальный номер из шести цифр, первая
Каждому фенотипу в OMIM присвоен уникальный номер из шести цифр, первая

Символ (*) перед номером указывает гены с известной последовательностью.
Символ (#) перед
Символ (*) перед номером указывает гены с известной последовательностью.
Символ (#) перед
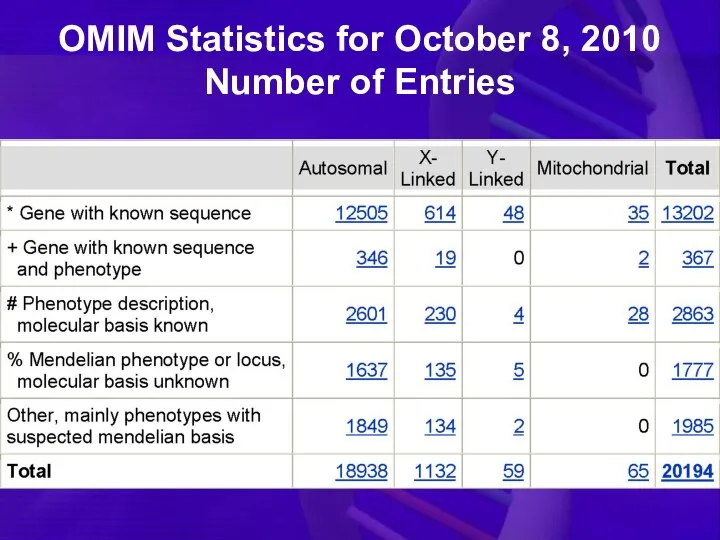
OMIM Statistics for October 8, 2010
Number of Entries
OMIM Statistics for October 8, 2010
Number of Entries
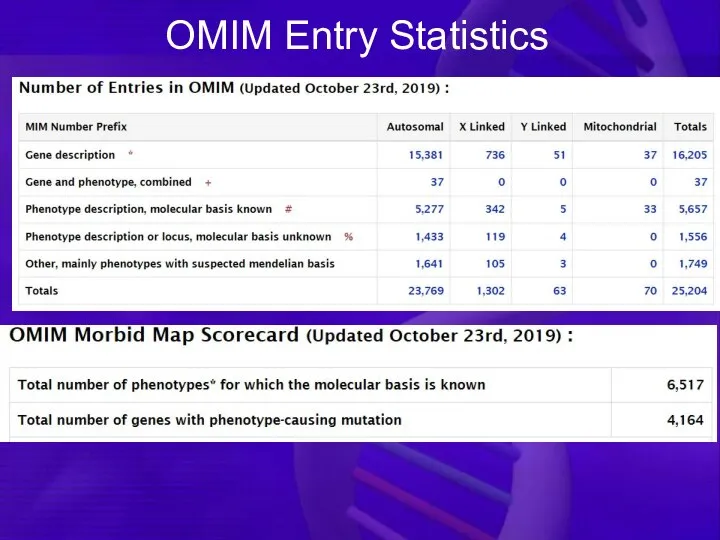
OMIM Entry Statistics
OMIM Entry Statistics

OMIM Entry Statistics
OMIM Entry Statistics
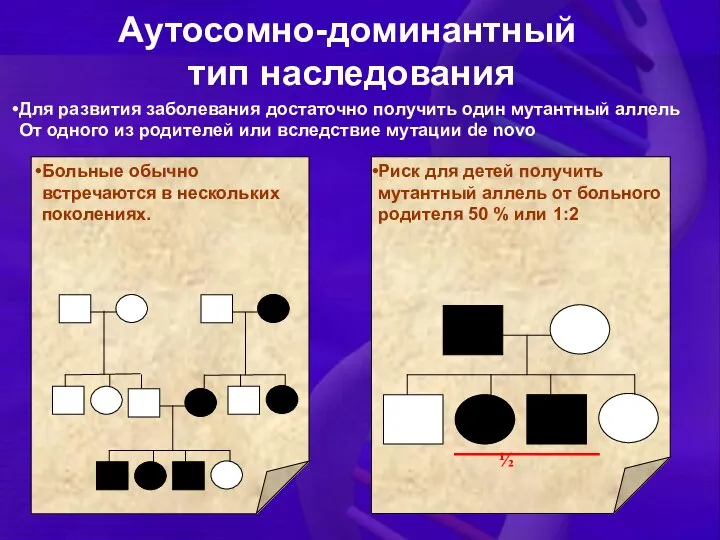
Для развития заболевания достаточно получить один мутантный аллель
От одного из родителей
Для развития заболевания достаточно получить один мутантный аллель
От одного из родителей
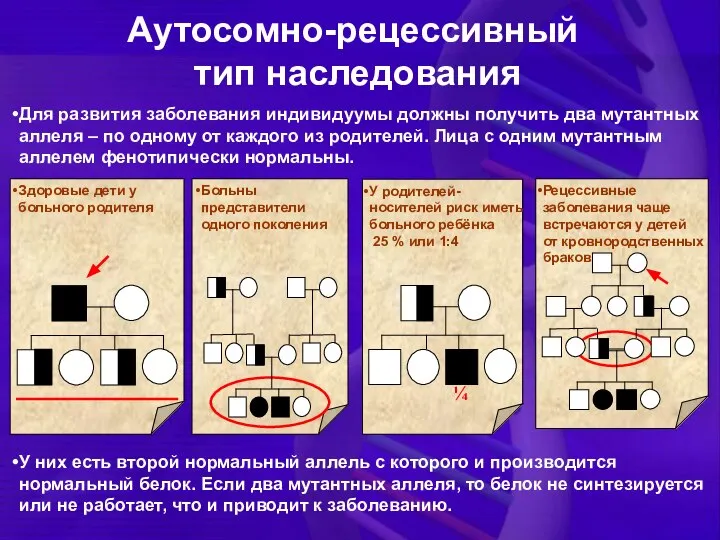
Для развития заболевания индивидуумы должны получить два мутантных аллеля – по
Для развития заболевания индивидуумы должны получить два мутантных аллеля – по
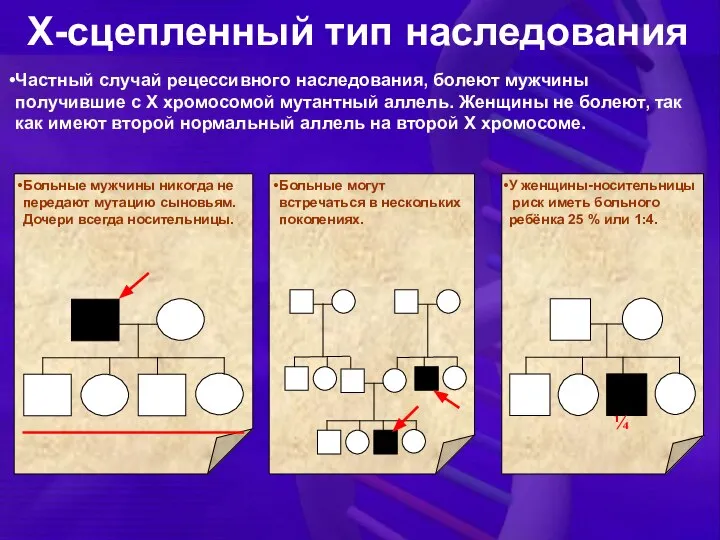
Частный случай рецессивного наследования, болеют мужчины получившие с Х хромосомой мутантный
Частный случай рецессивного наследования, болеют мужчины получившие с Х хромосомой мутантный
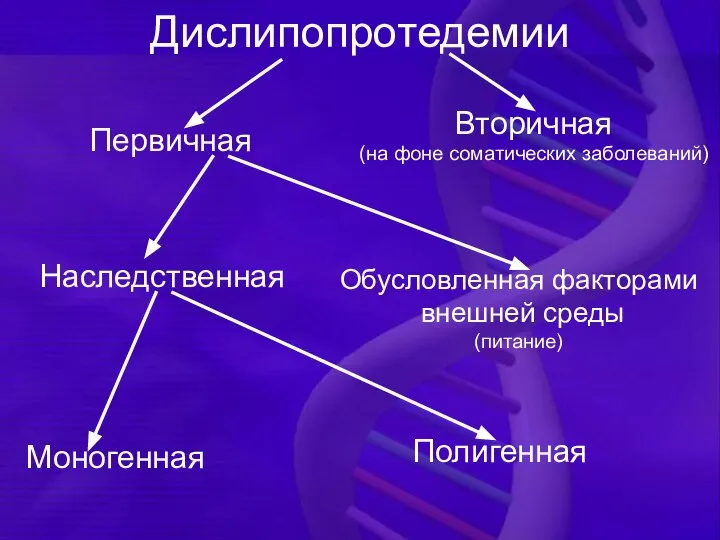
Дислипопротедемии
Первичная
Вторичная
(на фоне соматических заболеваний)
Наследственная
Обусловленная факторами
внешней среды
(питание)
Моногенная
Полигенная
Дислипопротедемии
Первичная
Вторичная
(на фоне соматических заболеваний)
Наследственная
Обусловленная факторами
внешней среды
(питание)
Моногенная
Полигенная

Моногенные болезни,
с нарушениями липидного обмена
Болезни накопления липидов (тезаурисмозы) - внутриклеточные
Моногенные болезни,
с нарушениями липидного обмена
Болезни накопления липидов (тезаурисмозы) - внутриклеточные

Семейная гиперхолестеринемия (143890)
Повышение уровня ЛПНП в крови
Частота гетерозиготной формы в
Семейная гиперхолестеринемия (143890)
Повышение уровня ЛПНП в крови
Частота гетерозиготной формы в
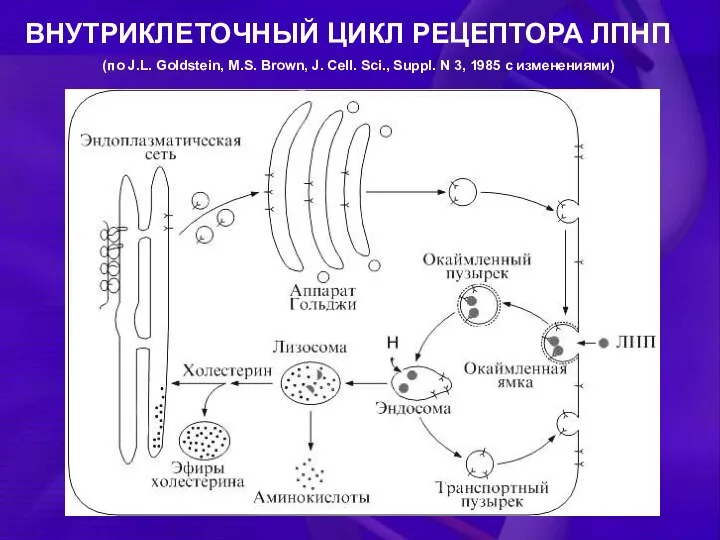
ВНУТРИКЛЕТОЧНЫЙ ЦИКЛ РЕЦЕПТОРА ЛПНП
(по J.L. Goldstein, M.S. Brown, J. Cell. Sci.,
ВНУТРИКЛЕТОЧНЫЙ ЦИКЛ РЕЦЕПТОРА ЛПНП
(по J.L. Goldstein, M.S. Brown, J. Cell. Sci.,

Рецептор ЛПНП синтезируется в виде предшественника с массой 120 кДа на
Рецептор ЛПНП синтезируется в виде предшественника с массой 120 кДа на
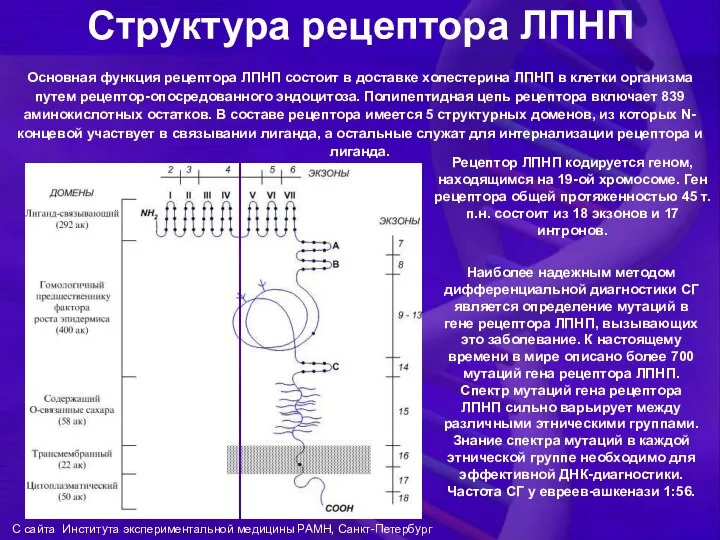
Структура рецептора ЛПНП
Наиболее надежным методом дифференциальной диагностики СГ является определение мутаций
Структура рецептора ЛПНП
Наиболее надежным методом дифференциальной диагностики СГ является определение мутаций
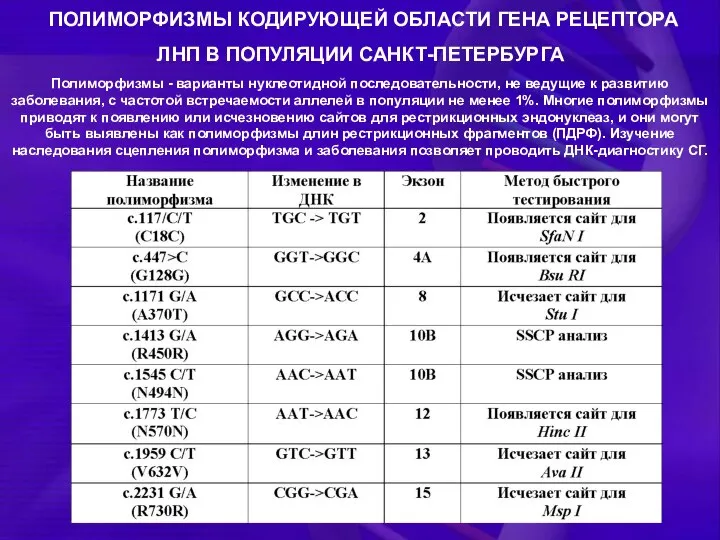
ПОЛИМОРФИЗМЫ КОДИРУЮЩЕЙ ОБЛАСТИ ГЕНА РЕЦЕПТОРА ЛНП В ПОПУЛЯЦИИ САНКТ-ПЕТЕРБУРГА
Полиморфизмы - варианты
ПОЛИМОРФИЗМЫ КОДИРУЮЩЕЙ ОБЛАСТИ ГЕНА РЕЦЕПТОРА ЛНП В ПОПУЛЯЦИИ САНКТ-ПЕТЕРБУРГА
Полиморфизмы - варианты

Семейная гиперхиломикронемия
Аккумуляция ХМ в плазме крови может быть обусловлена двумя
Семейная гиперхиломикронемия
Аккумуляция ХМ в плазме крови может быть обусловлена двумя

ГИПЕРЛИПОПРОТЕИНЕМИЯ ТИП 1 (+238600)
Синонимы
LIPOPROTEIN LIPASE DEFICIENCY
LPL DEFICIENCY
HYPERCHYLOMICRONEMIA, FAMILIAL
HYPERLIPEMIA, IDIOPATHIC, BURGER-GRUTZ
ГИПЕРЛИПОПРОТЕИНЕМИЯ ТИП 1 (+238600)
Синонимы
LIPOPROTEIN LIPASE DEFICIENCY
LPL DEFICIENCY
HYPERCHYLOMICRONEMIA, FAMILIAL
HYPERLIPEMIA, IDIOPATHIC, BURGER-GRUTZ

АПОЛИПОПРОТЕИН C-II ДЕФИЦИТ (207750)
Синонимы
HYPERLIPOPROTEINEMIA, TYPE IB
C-II ANAPOLIPOPROTEINEMIA
APOC2 DEFICIENCY
Ген локализован 19q13.2,
АПОЛИПОПРОТЕИН C-II ДЕФИЦИТ (207750)
Синонимы
HYPERLIPOPROTEINEMIA, TYPE IB
C-II ANAPOLIPOPROTEINEMIA
APOC2 DEFICIENCY
Ген локализован 19q13.2,

TANGIER DISEASE (205400); TGD
Синонимы
HIGH DENSITY LIPOPROTEIN DEFICIENCY, TYPE 1; HDLDT1
HIGH DENSITY
TANGIER DISEASE (205400); TGD
Синонимы
HIGH DENSITY LIPOPROTEIN DEFICIENCY, TYPE 1; HDLDT1
HIGH DENSITY

Абеталипопротеинемия (200100)
Генный локус 4q22-q24
Нарушение всасывания и транспорта жиров
Недостаточность или отсутствие бета-липопротеинов
Недостаточность
Абеталипопротеинемия (200100)
Генный локус 4q22-q24
Нарушение всасывания и транспорта жиров
Недостаточность или отсутствие бета-липопротеинов
Недостаточность

Ожирение
С-м Карпентера (201000)
С-м Альстрёма (203800)
С-м Барде-Бидля (209900)
С-м Бьерсона-Форсмана-Лемана (301900), Х-сцепленный рецессивный
Карликовость
Ожирение
С-м Карпентера (201000)
С-м Альстрёма (203800)
С-м Барде-Бидля (209900)
С-м Бьерсона-Форсмана-Лемана (301900), Х-сцепленный рецессивный
Карликовость

Липоатрофия, снижение или отсутствие подкожного жирового слоя
Синдром Вернера (277700)
Синдром Клиппеля-Треноне-Вебера (176670)
Синдром
Липоатрофия, снижение или отсутствие подкожного жирового слоя
Синдром Вернера (277700)
Синдром Клиппеля-Треноне-Вебера (176670)
Синдром

Прогерия,
синдром Гетчинсона-Гилфорда
Низкий рост и вес
Отсутствие подкожного жирового слоя
Тотальная алопеция
Маленькое лицо
Микрогнатия
Экзофтальм
Гиперхолестеринемия, ранний
Прогерия,
синдром Гетчинсона-Гилфорда
Низкий рост и вес
Отсутствие подкожного жирового слоя
Тотальная алопеция
Маленькое лицо
Микрогнатия
Экзофтальм
Гиперхолестеринемия, ранний

Синдром Вернера,
прогерия взрослых
Начало заболевания в 15-30 лет
Ювенильная катаракта
Преждевременное поседение и облысение
Склеродермия
Атрофия
Синдром Вернера,
прогерия взрослых
Начало заболевания в 15-30 лет
Ювенильная катаракта
Преждевременное поседение и облысение
Склеродермия
Атрофия

Липоматоз
Наследование аутосомно-доминантное.
Фенотипически проявляется безболезненными медленно растущими липомами. Необходимо дифференцировать с синдромом
Липоматоз
Наследование аутосомно-доминантное.
Фенотипически проявляется безболезненными медленно растущими липомами. Необходимо дифференцировать с синдромом
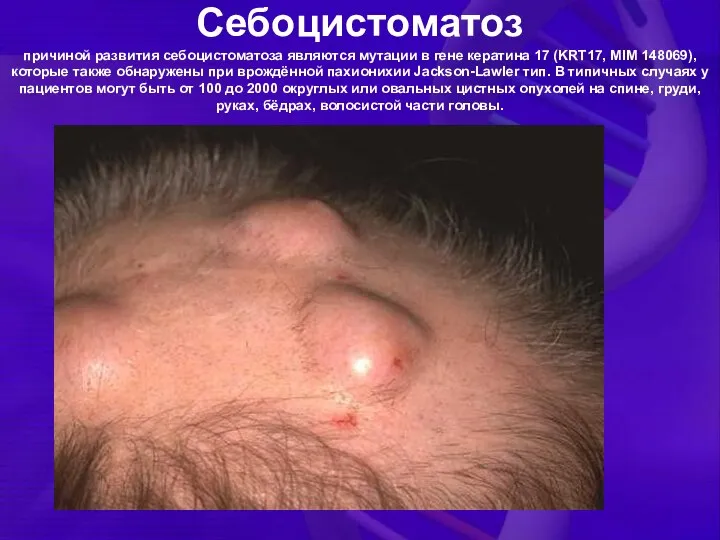
Себоцистоматоз
причиной развития себоцистоматоза являются мутации в гене кератина 17 (KRT17, MIM
Себоцистоматоз
причиной развития себоцистоматоза являются мутации в гене кератина 17 (KRT17, MIM

Генотерапия
Лечение СГХС. Резекция части печени, введение в клетки гена. Возврат клеток
Генотерапия
Лечение СГХС. Резекция части печени, введение в клетки гена. Возврат клеток
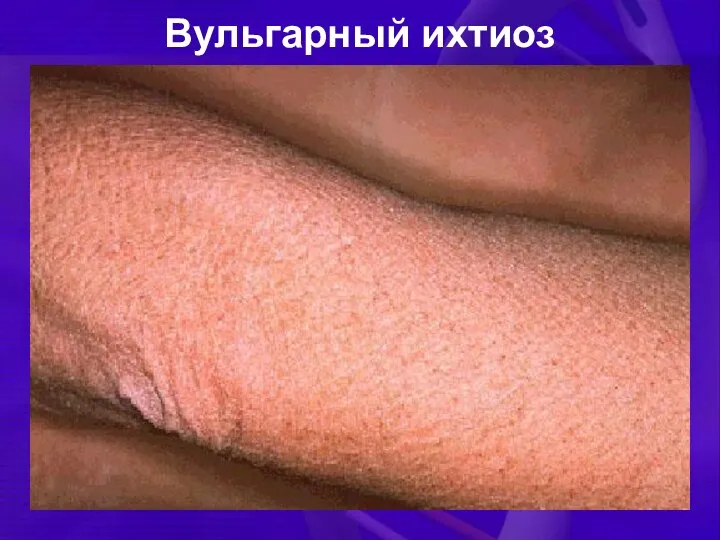
Вульгарный ихтиоз
Вульгарный ихтиоз
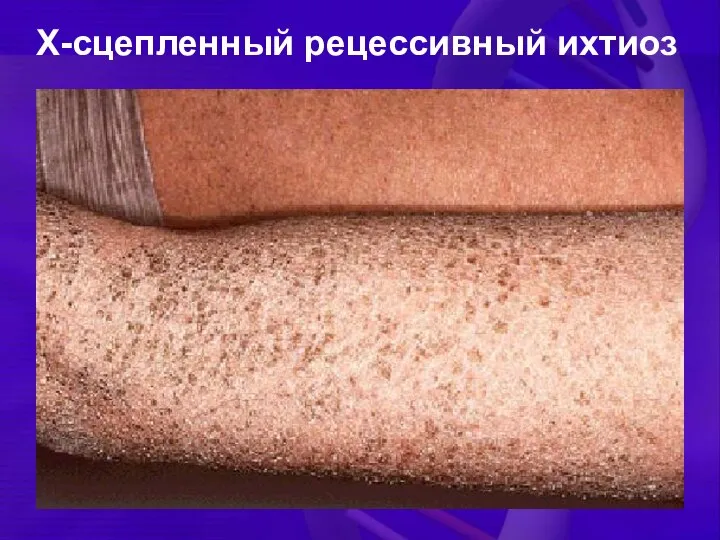
Х-сцепленный рецессивный ихтиоз
Х-сцепленный рецессивный ихтиоз

Классификация наследственных заболеваний обмена веществ
нарушения обмена аминокислот (фенилкетонурия и др.);
нарушения обмена
Классификация наследственных заболеваний обмена веществ
нарушения обмена аминокислот (фенилкетонурия и др.);
нарушения обмена

Признаки, требующие исключения НБО
В неонатальном периоде и на первом году жизни
Рвота,
Признаки, требующие исключения НБО
В неонатальном периоде и на первом году жизни
Рвота,

После первого года жизни
Умственная отсталость неясной этиологии.
УО с задержкой физического развития,
После первого года жизни
Умственная отсталость неясной этиологии.
УО с задержкой физического развития,

Симптомы и признаки болезней
обмена веществ у детей
Центральная нервная система
Летаргия, вялое сосание
Повышенная
Симптомы и признаки болезней
обмена веществ у детей
Центральная нервная система
Летаргия, вялое сосание
Повышенная

Органы дыхания и кровообращения
Апноэ
Тахипноэ
Респираторный дистресс
Нарушение функции печени
Желтуха
Гипокоагуляция
Гепатомегалия
Прочие нарушения
Патологический запах кожи или
Органы дыхания и кровообращения
Апноэ
Тахипноэ
Респираторный дистресс
Нарушение функции печени
Желтуха
Гипокоагуляция
Гепатомегалия
Прочие нарушения
Патологический запах кожи или

Врожденные дефекты метаболизма
А. Острое начало в неонатальный период
Заболевания встречаются редко, но
Врожденные дефекты метаболизма
А. Острое начало в неонатальный период
Заболевания встречаются редко, но

Б. Скрытое начало в период младенчества или детства
Некоторые заболевания неизлечимы и
Б. Скрытое начало в период младенчества или детства
Некоторые заболевания неизлечимы и

Болезни обмена веществ
у новорожденных
НАРУШЕНИЕ ОБМЕНА УГЛЕВОДОВ
Галактоземия
Дефицит фруктозо-1,6-дифосфатазы
Гликогенозы (1А и В типы)
НАРУШЕНИЕ
Болезни обмена веществ
у новорожденных
НАРУШЕНИЕ ОБМЕНА УГЛЕВОДОВ
Галактоземия
Дефицит фруктозо-1,6-дифосфатазы
Гликогенозы (1А и В типы)
НАРУШЕНИЕ

НАРУШЕНИЯ ОБМЕНА ПИРУВАТА И ТРАНСПОРТА ЭЛЕКТРОНОВ В ДЫХАТЕЛЬНОЙ ЦЕПИ
Дефицит пируваткарбоксилазы
Дефицит пируватдегидрогеназы
Дефицит
НАРУШЕНИЯ ОБМЕНА ПИРУВАТА И ТРАНСПОРТА ЭЛЕКТРОНОВ В ДЫХАТЕЛЬНОЙ ЦЕПИ
Дефицит пируваткарбоксилазы
Дефицит пируватдегидрогеназы
Дефицит

Заболевания, на которые проводится неонатальный скрининг в России
Фенилкетонурия
Врождённый гипотиреоз
Муковисцидоз
Галактоземия
Адреногенитальный синдром
Заболевания, на которые проводится неонатальный скрининг в России
Фенилкетонурия
Врождённый гипотиреоз
Муковисцидоз
Галактоземия
Адреногенитальный синдром
 ПОЛОЖЕНИЕ ЧЕЛОВЕКА В СИСТЕМЕ ЖИВОТНОГО МИРА
ПОЛОЖЕНИЕ ЧЕЛОВЕКА В СИСТЕМЕ ЖИВОТНОГО МИРА Всеядные птицы
Всеядные птицы Многообразие паукообразных
Многообразие паукообразных Методы исследования уровня экспрессии генов
Методы исследования уровня экспрессии генов Что такое весна. Загадки, раскраски, прописи
Что такое весна. Загадки, раскраски, прописи Биологическое действие ионизирующих излучений
Биологическое действие ионизирующих излучений Презентация на тему "МНОГООБРАЗИЕ ПРОСТЕЙШИХ" - скачать бесплатно презентации по Биологии
Презентация на тему "МНОГООБРАЗИЕ ПРОСТЕЙШИХ" - скачать бесплатно презентации по Биологии Заттардың тасымалдануы. Гуморальді және жасушалық иммунитетті салыстыру
Заттардың тасымалдануы. Гуморальді және жасушалық иммунитетті салыстыру Живое вещество биосферы и его функции
Живое вещество биосферы и его функции Презентация на тему Правила поведения в кабинете биологии и химии.
Презентация на тему Правила поведения в кабинете биологии и химии.  Скелет головы и туловища
Скелет головы и туловища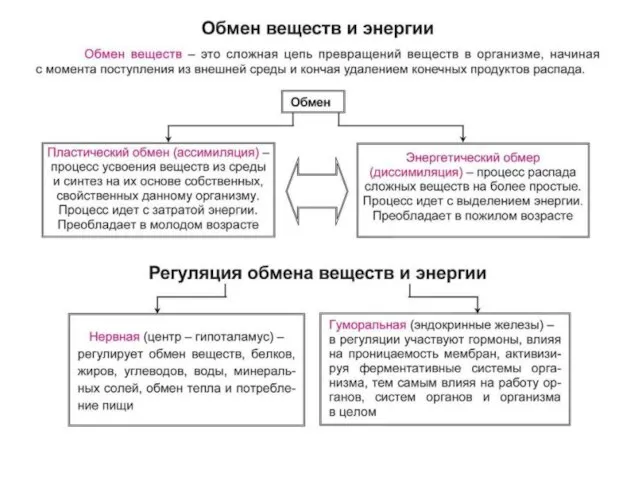 Обмен веществ и энергии
Обмен веществ и энергии Общие вопросы патогенеза эстетических дисфункций и особенности лечебных техник. Раздел №1
Общие вопросы патогенеза эстетических дисфункций и особенности лечебных техник. Раздел №1 Многообразие биогеоценозов (экосистем)
Многообразие биогеоценозов (экосистем) Торможение, его виды и значение
Торможение, его виды и значение Практическая работа №1. Анализ почвы пришкольного участка
Практическая работа №1. Анализ почвы пришкольного участка Definiţia, sistematizarea, importanţa şi rolul anatomiei animalelor domestice ca ştiinţă. (Tema 1)
Definiţia, sistematizarea, importanţa şi rolul anatomiei animalelor domestice ca ştiinţă. (Tema 1) Презентация на тему БИОЛОГИЯ РАСТЕНИЯ
Презентация на тему БИОЛОГИЯ РАСТЕНИЯ  Биоценоз Презентация по биологии
Биоценоз Презентация по биологии Цитология. Строение клетки по данным световой микроскопии
Цитология. Строение клетки по данным световой микроскопии Как помочь птицам зимой
Как помочь птицам зимой Необычные растения
Необычные растения Тайна дрожжей. (3 класс)
Тайна дрожжей. (3 класс) Порівняння обсягів і структури забруднення міст України
Порівняння обсягів і структури забруднення міст України  Презентация Ознакомление с окружающим миром, начальная школа
Презентация Ознакомление с окружающим миром, начальная школа Размножение. Типы размножения
Размножение. Типы размножения Задача Дерево Теслы
Задача Дерево Теслы Происхождение растений. Основные этапы развития растительного мира
Происхождение растений. Основные этапы развития растительного мира