- Химическая кинетика

Содержание
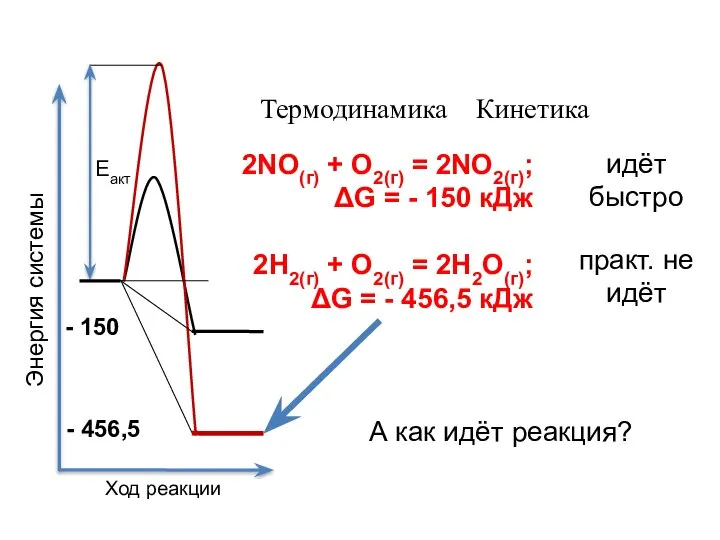
- 2. Еакт 2NO(г) + О2(г) = 2NO2(г); ΔG = - 150 кДж 2Н2(г) + О2(г) = 2Н2О(г);
- 3. Механизм химической реакции - совокупность элементарных стадий, из к-рых складывается хим. реакция Большинство р-ций осуществляется не
- 4. По числу ч-ц или мол-л, участвующих в элем. акте, судят о молекулярности реакции. 2А + В
- 5. Установление детального механизма хим. р-ции явл-ся сложной задачей и основано, в 1-ую очередь, на изучении скорости
- 6. при V=Const : где С – конц-ция. Влияние концентрации реагентов. Основной закон кинетики Давно известно, что
- 7. Математически ОЗК даётся в виде кинетическо-го ур-ния р-ции: ϑ = К·[A1]p·[A2]q····[Aℓ]r. К - константа скорости р-ции,
- 8. Для них кинетич. ур-ние р-ции выражает сущность основного постулата хим. кинетики – закона действия (действующих) масс
- 9. 3. если мех-зм простой (одна стадия), то ϑр-ции = К·[A]2·[В] если ϑ1 ϑр-ции = ϑ1 =
- 10. Влияние т-ры. Энергия активации процесса. Впервые количественная зав-сть скорости р-ции от т-ры была дана ≈ в
- 11. В чём причина зависимости? Впервые это сделал Аррениус (1889 г). Суть гипотезы Аррениуса: в эффективном столкновении
- 12. Величина Еакт зависит от природы в-в и мех-зма р-ции (от 0 до 500 кДж/моль). Преодолеть барьер
- 13. закон Максвелла-Больцмана В газовой смеси мол-лы обладают различной энергией. Наибольшая часть мол-л газа имеет энергию, близкую
- 15. Влияние катализатора. Понятие о катализе Поскольку кат-р после р-ции остаётся в неизменном состоянии и количестве, то
- 16. чем меньше энергия акт-ции (Е**акт А + В → АВ Мех-зм действия кат-ра: А + К
- 17. ϑ ϑпр ϑобр ϑпр=ϑобр t Рис. Изменение скорости прямой (ϑ1) и обратной (ϑ2) реакций во времени
- 18. МОДЕЛЬ ХИМИЧЕСКОГО РАВНОВЕСИЯ
- 19. Смещение равновесия Принцип Ле Шателье Если на систему, находящуюся в состоянии равновесия, оказано внешнее воздействие, то
- 20. Основные факторы, влияющие на нарушение равновесия: концентрация веществ давление температура Процесс изменения конц-ций, вызванный нарушением равновесия,
- 21. Влияние температуры Ур-ние изотермы хим. р-ции (Я.Вант-Гофф) для стандартных условий имеет вид: ΔG°Т = –RTlnKp(T). При
- 22. Закономерности сдвига равновесия в химических системах есть частный случай общего принципа поведения равновесных систем. Это принцип
- 24. Скачать презентацию
Еакт
2NO(г) + О2(г) = 2NO2(г);
ΔG = - 150 кДж
2Н2(г) +
Еакт
2NO(г) + О2(г) = 2NO2(г);
ΔG = - 150 кДж
2Н2(г) +

Механизм химической реакции
- совокупность элементарных стадий, из к-рых складывается хим. реакция
Большинство
Механизм химической реакции
- совокупность элементарных стадий, из к-рых складывается хим. реакция
Большинство

По числу ч-ц или мол-л, участвующих в элем. акте, судят о
По числу ч-ц или мол-л, участвующих в элем. акте, судят о
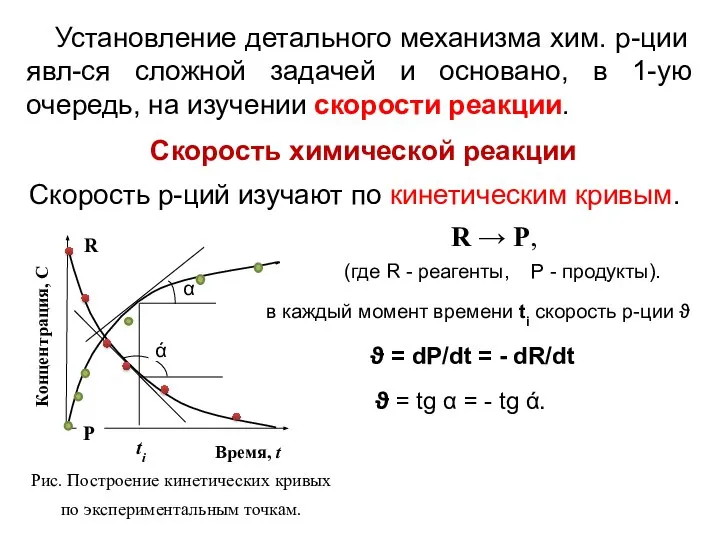
Установление детального механизма хим. р-ции явл-ся сложной задачей и основано, в
Установление детального механизма хим. р-ции явл-ся сложной задачей и основано, в

при V=Const :
где С – конц-ция.
Влияние концентрации реагентов.
Основной закон кинетики
Давно
при V=Const :
где С – конц-ция.
Влияние концентрации реагентов.
Основной закон кинетики
Давно
![Математически ОЗК даётся в виде кинетическо-го ур-ния р-ции: ϑ = К·[A1]p·[A2]q····[Aℓ]r.](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1397112/slide-6.jpg)
Математически ОЗК даётся в виде кинетическо-го ур-ния р-ции:
ϑ = К·[A1]p·[A2]q····[Aℓ]r.
К -
Математически ОЗК даётся в виде кинетическо-го ур-ния р-ции:
ϑ = К·[A1]p·[A2]q····[Aℓ]r.
К -

Для них кинетич. ур-ние р-ции выражает сущность основного постулата хим. кинетики
Для них кинетич. ур-ние р-ции выражает сущность основного постулата хим. кинетики
![3. если мех-зм простой (одна стадия), то ϑр-ции = К·[A]2·[В] если](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/1397112/slide-8.jpg)
3. если мех-зм простой (одна стадия), то
ϑр-ции = К·[A]2·[В]
если ϑ1
3. если мех-зм простой (одна стадия), то
ϑр-ции = К·[A]2·[В]
если ϑ1

Влияние т-ры. Энергия активации процесса.
Впервые количественная зав-сть скорости р-ции от т-ры
Влияние т-ры. Энергия активации процесса.
Впервые количественная зав-сть скорости р-ции от т-ры

В чём причина зависимости?
Впервые это сделал Аррениус (1889 г).
Суть
В чём причина зависимости?
Впервые это сделал Аррениус (1889 г).
Суть
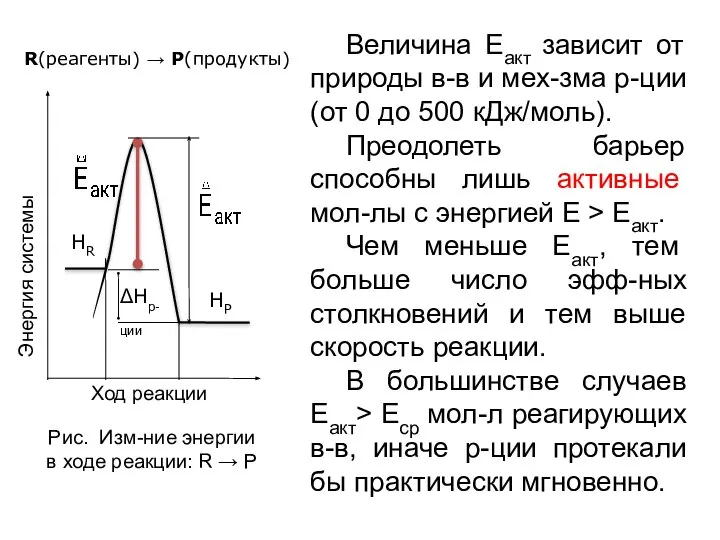
Величина Еакт зависит от природы в-в и мех-зма р-ции (от 0
Величина Еакт зависит от природы в-в и мех-зма р-ции (от 0
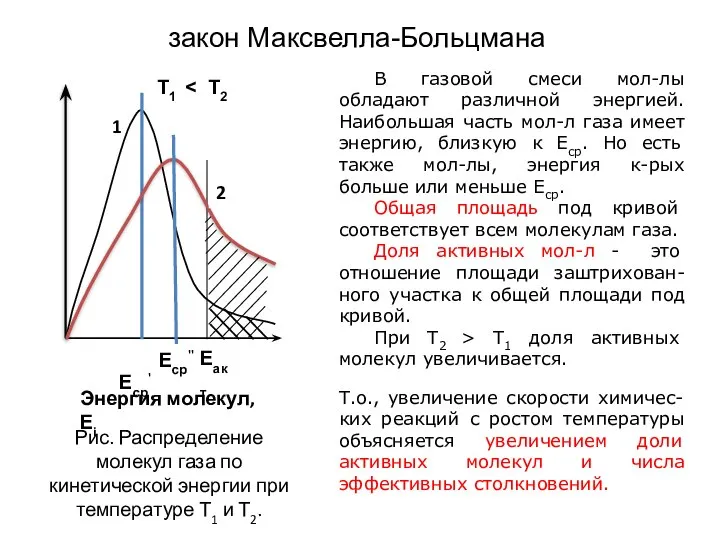
закон Максвелла-Больцмана
В газовой смеси мол-лы обладают различной энергией. Наибольшая часть мол-л
закон Максвелла-Больцмана
В газовой смеси мол-лы обладают различной энергией. Наибольшая часть мол-л


Влияние катализатора. Понятие о катализе
Поскольку кат-р после р-ции остаётся в неизменном
Влияние катализатора. Понятие о катализе
Поскольку кат-р после р-ции остаётся в неизменном
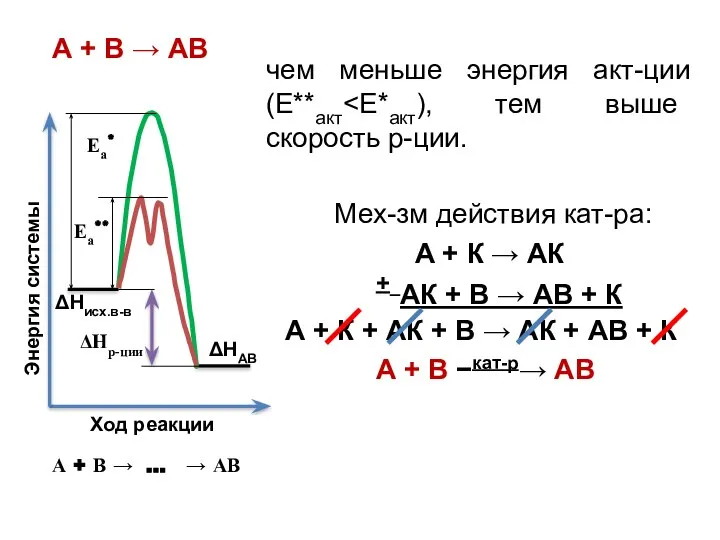
чем меньше энергия акт-ции (Е**акт<Е*акт), тем выше скорость р-ции.
А + В
чем меньше энергия акт-ции (Е**акт<Е*акт), тем выше скорость р-ции.
А + В
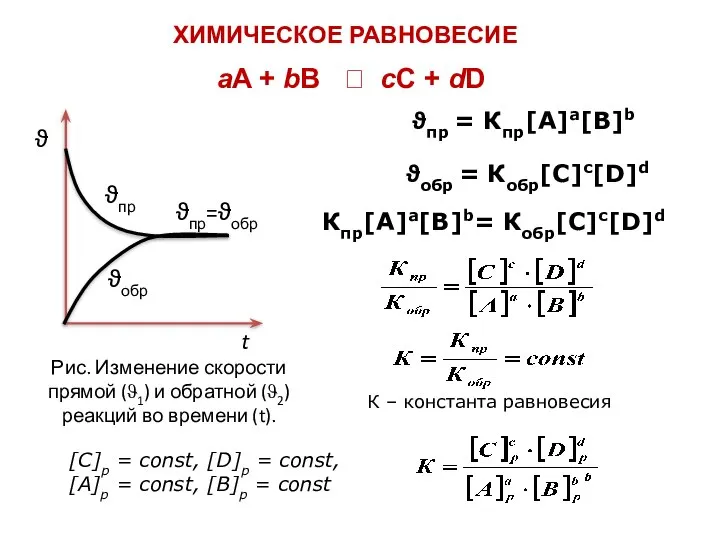
ϑ
ϑпр
ϑобр
ϑпр=ϑобр
t
Рис. Изменение скорости прямой (ϑ1) и обратной (ϑ2) реакций во времени
ϑ
ϑпр
ϑобр
ϑпр=ϑобр
t
Рис. Изменение скорости прямой (ϑ1) и обратной (ϑ2) реакций во времени
МОДЕЛЬ ХИМИЧЕСКОГО РАВНОВЕСИЯ
МОДЕЛЬ ХИМИЧЕСКОГО РАВНОВЕСИЯ
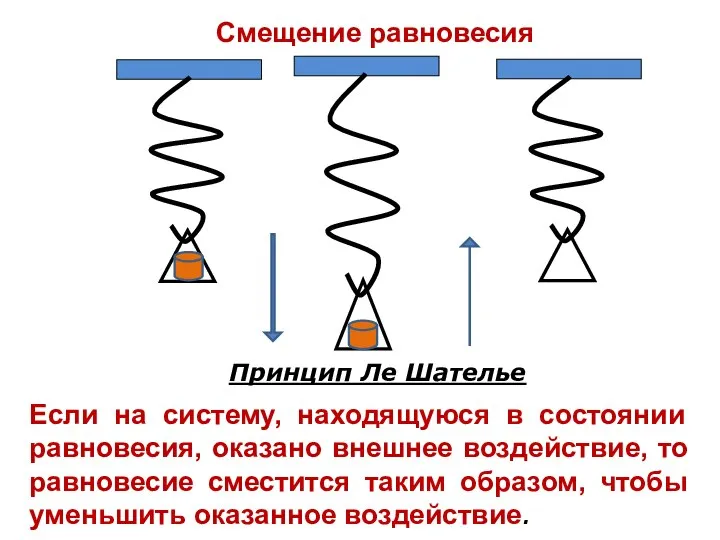
Смещение равновесия
Принцип Ле Шателье
Если на систему, находящуюся в состоянии равновесия,
Смещение равновесия
Принцип Ле Шателье
Если на систему, находящуюся в состоянии равновесия,
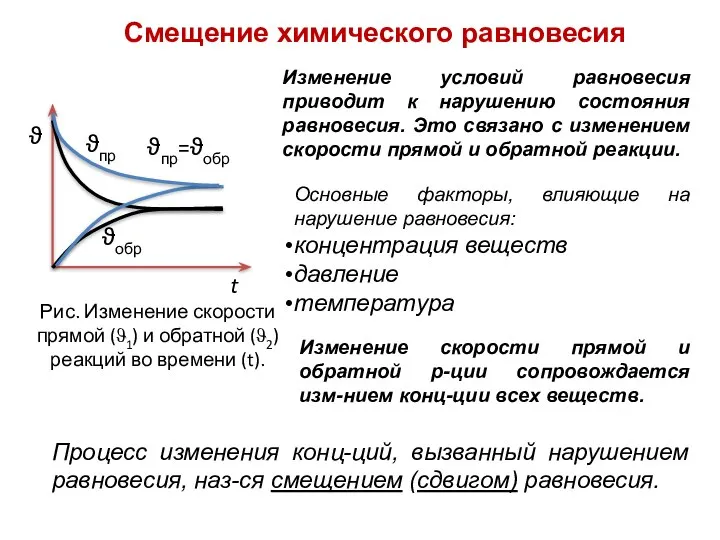
Основные факторы, влияющие на нарушение равновесия:
концентрация веществ
давление
температура
Процесс изменения конц-ций, вызванный нарушением
Основные факторы, влияющие на нарушение равновесия:
концентрация веществ
давление
температура
Процесс изменения конц-ций, вызванный нарушением
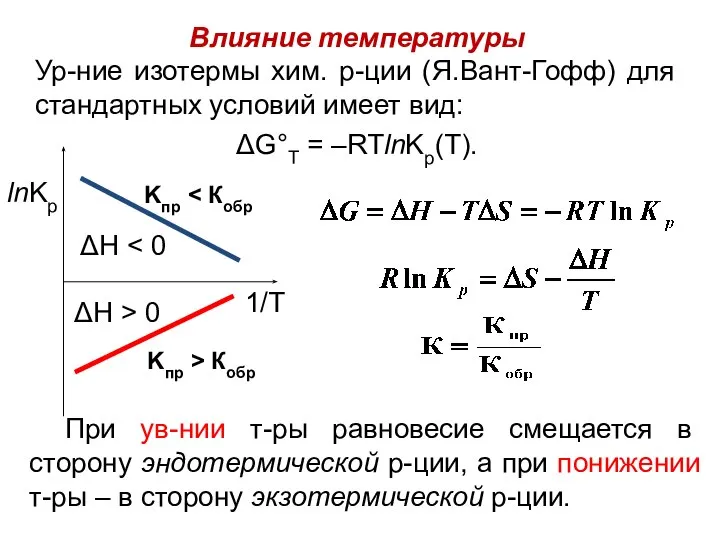
Влияние температуры
Ур-ние изотермы хим. р-ции (Я.Вант-Гофф) для стандартных условий имеет вид:
Влияние температуры
Ур-ние изотермы хим. р-ции (Я.Вант-Гофф) для стандартных условий имеет вид:

Закономерности сдвига равновесия в химических системах есть частный случай общего принципа
Закономерности сдвига равновесия в химических системах есть частный случай общего принципа
 Презентация по Химии "Моносахариды" - скачать смотреть
Презентация по Химии "Моносахариды" - скачать смотреть  Энергетический обмен. Биологическое окисление
Энергетический обмен. Биологическое окисление Характеристика металу Sr
Характеристика металу Sr Технология производства простых полиэфиров
Технология производства простых полиэфиров Метаморфогенная серия. Группа регионального метаморфизма
Метаморфогенная серия. Группа регионального метаморфизма Азотная кислота
Азотная кислота Жиры. 9 класс
Жиры. 9 класс Презентация Спирты
Презентация Спирты Полярные электронные эффекты в органических соединениях
Полярные электронные эффекты в органических соединениях Щелочные и щелочноземельные металлы и их роль в организме человека
Щелочные и щелочноземельные металлы и их роль в организме человека Производство серной кислоты контактным способом
Производство серной кислоты контактным способом Презентация по Химии "Строение электронных оболочек атомов" - скачать смотреть
Презентация по Химии "Строение электронных оболочек атомов" - скачать смотреть  Тема урока Агрегатные состояния вещества. Плавление и отвердевание кристаллических тел.
Тема урока Агрегатные состояния вещества. Плавление и отвердевание кристаллических тел. Региональная металлогения
Региональная металлогения ГАЛОГЕНЫ И ИХ СОЕДИНЕНИЯ
ГАЛОГЕНЫ И ИХ СОЕДИНЕНИЯ  Биохимия нервной ткани. (Лекция 23)
Биохимия нервной ткани. (Лекция 23) Комплексные соединения
Комплексные соединения Химические свойства оснований. С какими из перечисленных веществ может реагировать данное вещество
Химические свойства оснований. С какими из перечисленных веществ может реагировать данное вещество Углерод Гончаров Никита 9 «Г» класс
Углерод Гончаров Никита 9 «Г» класс Резина и каучук
Резина и каучук Лаборатория мирового уровня в области термического анализа и физико-химии процессов тепловых методов добычи
Лаборатория мирового уровня в области термического анализа и физико-химии процессов тепловых методов добычи Выращивание кристаллов в домашних условиях
Выращивание кристаллов в домашних условиях Физико-химия полимеров и их растворов
Физико-химия полимеров и их растворов 10 самых смертельно опасных камней и минералов
10 самых смертельно опасных камней и минералов Содержание химических элементов в организме. Макро- и микроэлементы
Содержание химических элементов в организме. Макро- и микроэлементы Аммиак
Аммиак Тяжёлая вода
Тяжёлая вода Растворы электролитов
Растворы электролитов