- Наследственные митохондриальные болезни

Содержание
- 2. Общие сведения о нехромосомной наследственности Эта наследственность не подчиняется законам Менделя Все зеленые Реципрокные скрещивания Все
- 3. Строение митохондрий 37 генов
- 4. Митохондриальная ДНК человека 16659 пн L (Light) — легкая, богата цитозином, содержит 9 генов: ND6, 8
- 5. Причины и особенности заболеваний Основные причины: 1. Мутации митохондриальных генов 2. Мутации генов яДНК необходимых для
- 6. Типы наследования митохондриальных заболеваний Материнское (митохондриальное или цитоплазматическое) Спорадические случаи Менделевское наследование Синдромы: MELAS, MERRF, NARP,
- 7. Заболевания наследуемые по цитоплазматическому типу
- 8. Синдром MELAS митохондриальная энцефаломиопатия (недостаточное поступление крови к мозгу, образование отеков), лактатацидоз (очень много молочной кислоты
- 9. Синдром MERRF Точковые мутации мтДНК в гене, кодирующем тРНК лизина A8344G, на долю которой приходится свыше
- 10. Болезнь LHON (Лебера) Наиболее частая причина — мутация в нуклеотиде 11778 мтДНК. Нуклеотид находится в пределах
- 11. Спорадические случаи Синдром Пирсона Бледность, вялость, сонливость. Нарушения кроветворения (прежде всего выработки эритроцитов). Отставание в развитии.
- 12. Заболевания с Мендлеевским типом наследования Болеют 50 % детей, рожденных от больного мужчины или больной женщины.
- 13. Атаксия Фридрейха Аутосомно-рецессивный тип наследования. Мутацией является экспансия GAA тринуклеотидных повторов (200-900) в гене фратаксина (9q13).
- 14. Синдром Альперса Мутации ядерного гена POLG1, кодирующего митохондриальную полимеразу 2–4 год Микроцефалия; Задержка психического и физического
- 15. Синдром Лея (Ли) Точечные мутации мтДНК; либо ядерные мутации или других генов OXPHOS. 1–2 год это
- 17. Скачать презентацию

Общие сведения о нехромосомной наследственности
Эта наследственность не подчиняется законам Менделя
Все зеленые
Реципрокные
Общие сведения о нехромосомной наследственности
Эта наследственность не подчиняется законам Менделя
Все зеленые
Реципрокные
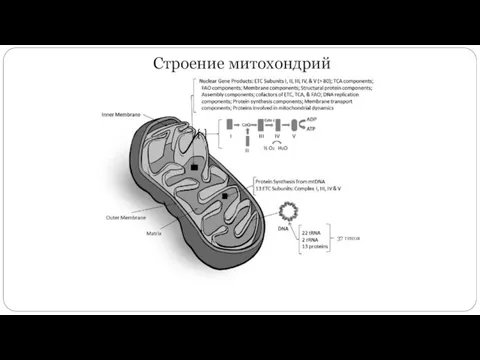
Строение митохондрий
37 генов
Строение митохондрий
37 генов

Митохондриальная ДНК человека
16659 пн
L (Light) — легкая, богата цитозином,
содержит 9
Митохондриальная ДНК человека
16659 пн
L (Light) — легкая, богата цитозином,
содержит 9
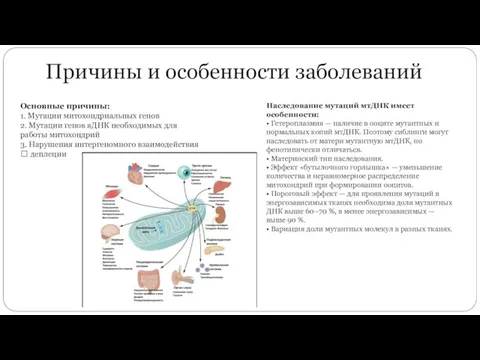
Причины и особенности заболеваний
Основные причины:
1. Мутации митохондриальных генов
2. Мутации генов
Причины и особенности заболеваний
Основные причины:
1. Мутации митохондриальных генов
2. Мутации генов

Типы наследования митохондриальных заболеваний
Материнское (митохондриальное или цитоплазматическое)
Спорадические случаи
Менделевское наследование
Синдромы:
MELAS,
MERRF,
Типы наследования митохондриальных заболеваний
Материнское (митохондриальное или цитоплазматическое)
Спорадические случаи
Менделевское наследование
Синдромы:
MELAS,
MERRF,

Заболевания наследуемые по цитоплазматическому типу
Заболевания наследуемые по цитоплазматическому типу

Синдром MELAS
митохондриальная
энцефаломиопатия (недостаточное поступление крови к мозгу, образование отеков), лактатацидоз (очень
Синдром MELAS
митохондриальная энцефаломиопатия (недостаточное поступление крови к мозгу, образование отеков), лактатацидоз (очень
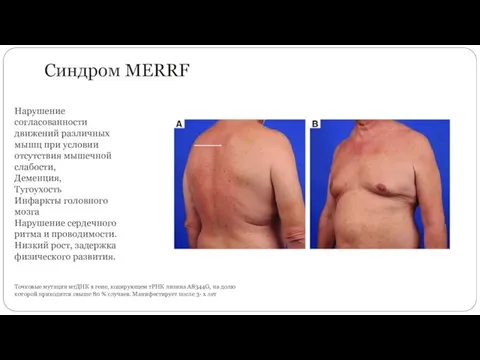
Синдром MERRF
Точковые мутации мтДНК в гене, кодирующем тРНК лизина A8344G, на
Синдром MERRF
Точковые мутации мтДНК в гене, кодирующем тРНК лизина A8344G, на

Болезнь LHON (Лебера)
Наиболее частая причина — мутация в нуклеотиде 11778 мтДНК.
Болезнь LHON (Лебера)
Наиболее частая причина — мутация в нуклеотиде 11778 мтДНК.

Спорадические случаи
Синдром Пирсона
Бледность, вялость, сонливость. Нарушения кроветворения (прежде всего выработки эритроцитов).
Спорадические случаи
Синдром Пирсона
Бледность, вялость, сонливость. Нарушения кроветворения (прежде всего выработки эритроцитов).

Заболевания с Мендлеевским типом наследования
Болеют 50 % детей, рожденных от больного
Заболевания с Мендлеевским типом наследования
Болеют 50 % детей, рожденных от больного
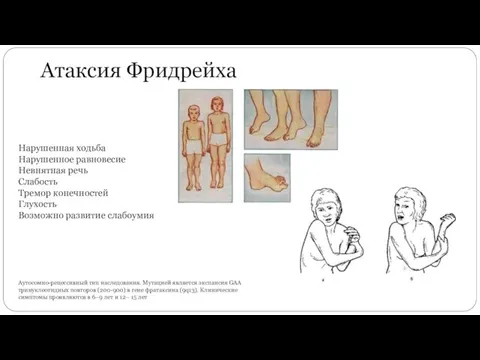
Атаксия Фридрейха
Аутосомно-рецессивный тип наследования. Мутацией является экспансия GAA тринуклеотидных повторов (200-900)
Атаксия Фридрейха
Аутосомно-рецессивный тип наследования. Мутацией является экспансия GAA тринуклеотидных повторов (200-900)

Синдром Альперса
Мутации ядерного гена POLG1, кодирующего митохондриальную полимеразу 2–4 год
Микроцефалия;
Задержка
Синдром Альперса
Мутации ядерного гена POLG1, кодирующего митохондриальную полимеразу 2–4 год
Микроцефалия;
Задержка

Синдром Лея (Ли)
Точечные мутации мтДНК; либо ядерные мутации или других генов
Синдром Лея (Ли)
Точечные мутации мтДНК; либо ядерные мутации или других генов
 Структура и физиология плодных оболочек плаценты
Структура и физиология плодных оболочек плаценты Сахарный диабет и его поздние осложнения
Сахарный диабет и его поздние осложнения Патофизиология почек. Острая и хроническая почечная недостаточность. Гломерулонефрит. Нефротический синдром
Патофизиология почек. Острая и хроническая почечная недостаточность. Гломерулонефрит. Нефротический синдром Кишечные инфекции: эпидемиология, клиника и профилактика
Кишечные инфекции: эпидемиология, клиника и профилактика Аномалии прикуса
Аномалии прикуса Влияние фастфуда на организм человека
Влияние фастфуда на организм человека СП при пороках сердца
СП при пороках сердца ХОБЛ – как медико-социальная проблема
ХОБЛ – как медико-социальная проблема Артықшылықты режим. Науқастардың санаттары
Артықшылықты режим. Науқастардың санаттары Наука о весе тела и питании человека
Наука о весе тела и питании человека Направление и основные ветви лучевого нерва
Направление и основные ветви лучевого нерва Аралығында гидроцефалия диагнозымен ауратын балаларға жасалынатын шундтау операциясынан нейроэндоскопия операциясының тиімділігі
Аралығында гидроцефалия диагнозымен ауратын балаларға жасалынатын шундтау операциясынан нейроэндоскопия операциясының тиімділігі Неотложные состояния у детей
Неотложные состояния у детей Обзор сердечно-сосудистой системы. Общие принципы строение кровеносного и лимфатического сосудистых русел. Лекция № 25
Обзор сердечно-сосудистой системы. Общие принципы строение кровеносного и лимфатического сосудистых русел. Лекция № 25 Травматические повреждения позвоночника
Травматические повреждения позвоночника ВИЧ и СПИД
ВИЧ и СПИД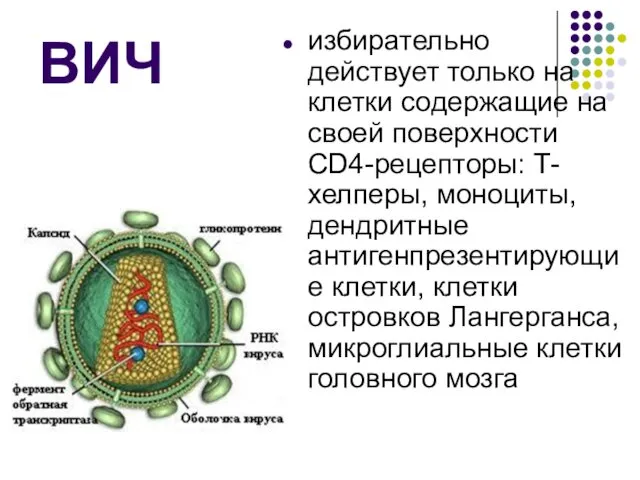 ВИЧ-инфекция
ВИЧ-инфекция Араб халифаттарының медицинасы. Абу Али ибн Сина. Дәрігерлік ғылым қағидалары
Араб халифаттарының медицинасы. Абу Али ибн Сина. Дәрігерлік ғылым қағидалары Познавательные процессы
Познавательные процессы Профилактика дыхательной системы
Профилактика дыхательной системы SARS-CoV-2. Тяжёлый острый респираторный синдром – коронавирус 2 COVID – 19
SARS-CoV-2. Тяжёлый острый респираторный синдром – коронавирус 2 COVID – 19 Международный день врача
Международный день врача Лимфомы. Миеломная болезнь
Лимфомы. Миеломная болезнь Методы нейровизуализации при ишемических инсультах. Диагностическая значимость КТ, МРТ
Методы нейровизуализации при ишемических инсультах. Диагностическая значимость КТ, МРТ Сесилия Сандерс «Хоспистер анасы»
Сесилия Сандерс «Хоспистер анасы» Патофизиология углеводного обмена
Патофизиология углеводного обмена Предмет и методы невропатологии. Связь невропатологии и коррекционной педагогики
Предмет и методы невропатологии. Связь невропатологии и коррекционной педагогики Диссеминированный туберкулез легких
Диссеминированный туберкулез легких