- ЭНЗИМОПАТИИ (наследственные болезни связанные с нарушением обмена веществ)

Содержание
- 2. Процессы обмена веществ в клетке находятся под контролем: Нервной и эндокринной регуляции, обеспечивающих согласование обменных процессов
- 3. -Полная блокада (выключение) синтеза фермента; -Снижения активности фермента; -Нарушения других систем или биохимических реакций, от которых
- 4. ВЫЯВЛЕНИЕ И ДИАГНОЗ Фенилкетонурия и галактоземия (нарушение углеводного обмена), могут быть определены путем анализа крови, взятой
- 5. Внутриутробное развитие ребенка
- 7. Наследственные нарушения аминокислот (энзимопатии) - — группа заболеваний, обусловленных дефектами ферментов, участвующих в их обмене. Описано
- 8. Подавляющее большинство этих болезней наследуется аутосомно-рецессивно. Отдельные формы заболеваний передаются с Х-хромосомой. При большинстве нарушений аминокислотного
- 9. 1. Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых аминокислот. 3. Назначение препаратов,
- 10. Примеры энзимопатий
- 11. Фенилкетонурия (ФКУ) Впервые описал A. Foiling в 1934 году. Частота встречаемости в России — 1:10000. ФКУ
- 12. Патогенез Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением фенилаланина в тирозин. В результате этого концентрация
- 13. Гиперкинезы, нарушения мышечного тонуса и координации. 2. 25—50% больных страдают припадками. 3. Нарушения пигментации (светлый цвет
- 14. Ребенок 2-х лет с фенилкетонурией
- 15. Психическое развитие Отставание в психическом развитии становится заметным во втором полугодии жизни и прогрессирует в течение
- 16. Лечение Диета с резким ограничением фенилаланина с 2— 3 -месячного возраста и соблюдение ее в течение
- 17. ОРГАНИЧЕСКИЕ АЦИДЕМИИ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ НЕРВНО-ПСИХИЧЕСКОГО РАЗВИТИЯ Эти нарушения метаболизма органических кислот (продуктов обмена аминокислот, углеводов, липидов,
- 18. В 1967 году (Budd M. А. et al.) впервые была уточнена природа одной из ацидемий (изовалериановой).
- 19. Первичные симптомы: - респираторный и нейродистресс-синдромы, - припадки, - рвота, отказ от еды, нарушение стула, обезвоживание.
- 20. Вторичные нарушения: - отставание психического и моторного развития, -пирамидная симптоматика, -расстройства координации, -судороги. Особые симптомы отмечаются
- 21. Лечение ограничение белка; высокие дозах витаминов; дополнительное введение Л-карнитина и глицина.
- 22. Галактоземия Описана в 1908 году, однако дефект обмена, ее обуславливающий, был открыт лишь в 1956 году.
- 23. Патогенез Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар) не усваивается, а промежуточный продукт
- 24. Клиника Проявляется вскоре после рождения у ребенка: отказом от пищи, поносом, рвотой, непереносимостью голода, падением массы
- 25. Лечение Безмолочная диета
- 26. МУКОПОЛИСАХАРИДО3 1 Н (СИНДРОМ ГУРЛЕРА) Описан G. Gurler в 1919 году. Встречается с частотой — 1:
- 27. Клиника Проявляется на первом году жизни. Внешний вид больных — увеличенная голова, выдающиеся лобные бугры, почти
- 28. Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный дефект усугубляется, затем происходит потеря приобретенных
- 29. Патогенез Отложение мукополисахаридов в соединительной ткани печени, селезенки и других тканях. Накопление мукополисахаридов в хрящах нарушает
- 30. Болезнь Гирке Нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях организма. Связано с недостаточностью
- 31. Альбинизм При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене) образуется кожный
- 32. Алкаптонурия Заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты — промежуточного продукта обмена
- 33. Гиперхолестеринемия Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном находится)
- 35. Скачать презентацию

Процессы обмена веществ в клетке находятся под контролем:
Нервной
Процессы обмена веществ в клетке находятся под контролем:
Нервной

-Полная блокада (выключение) синтеза фермента;
-Снижения активности фермента;
-Нарушения других систем
-Полная блокада (выключение) синтеза фермента; -Снижения активности фермента; -Нарушения других систем

ВЫЯВЛЕНИЕ И ДИАГНОЗ
Фенилкетонурия и галактоземия (нарушение углеводного обмена), могут быть определены путем
ВЫЯВЛЕНИЕ И ДИАГНОЗ
Фенилкетонурия и галактоземия (нарушение углеводного обмена), могут быть определены путем
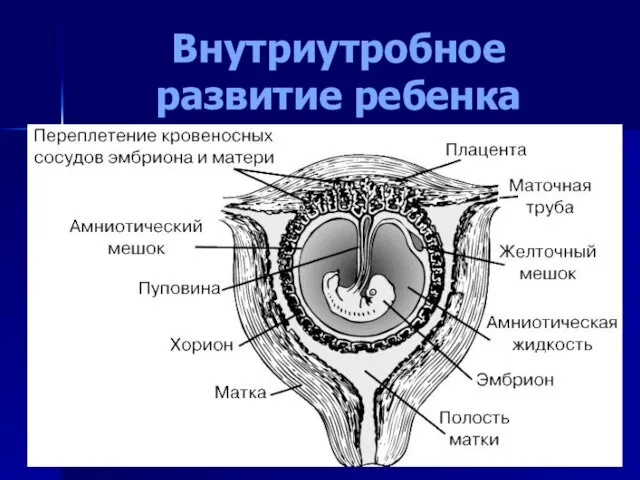
Внутриутробное развитие ребенка
Внутриутробное развитие ребенка

Наследственные нарушения аминокислот (энзимопатии) -
— группа заболеваний, обусловленных дефектами
Наследственные нарушения аминокислот (энзимопатии) -
— группа заболеваний, обусловленных дефектами

Подавляющее большинство этих болезней наследуется аутосомно-рецессивно.
Отдельные формы заболеваний передаются с
Подавляющее большинство этих болезней наследуется аутосомно-рецессивно. Отдельные формы заболеваний передаются с

1. Ограничение в диете белка и соответствующей аминокислоты.
2. Дополнительное назначение незаменимых
1. Ограничение в диете белка и соответствующей аминокислоты. 2. Дополнительное назначение незаменимых

Примеры энзимопатий

Фенилкетонурия
(ФКУ)
Впервые описал A. Foiling в 1934 году.
Частота встречаемости в
Фенилкетонурия
(ФКУ)
Впервые описал A. Foiling в 1934 году.
Частота встречаемости в

Патогенез
Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением фенилаланина
Патогенез
Поражение ЦНС вызывается недостаточностью фермента гидроксилазы-4-фенилаланина, управляющего превращением фенилаланина

Гиперкинезы, нарушения мышечного тонуса и координации.
2. 25—50% больных страдают
Гиперкинезы, нарушения мышечного тонуса и координации. 2. 25—50% больных страдают
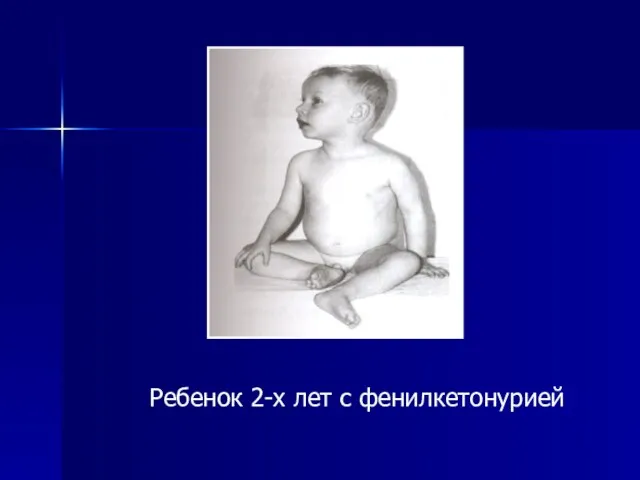
Ребенок 2-х лет с фенилкетонурией
Ребенок 2-х лет с фенилкетонурией

Психическое развитие
Отставание в психическом развитии становится заметным во втором полугодии
Психическое развитие
Отставание в психическом развитии становится заметным во втором полугодии

Лечение
Диета с резким ограничением фенилаланина с 2— 3 -месячного возраста и
Лечение
Диета с резким ограничением фенилаланина с 2— 3 -месячного возраста и

ОРГАНИЧЕСКИЕ АЦИДЕМИИ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ
НЕРВНО-ПСИХИЧЕСКОГО РАЗВИТИЯ
Эти нарушения метаболизма органических
ОРГАНИЧЕСКИЕ АЦИДЕМИИ, СОПРОВОЖДАЮЩИЕСЯ НАРУШЕНИЕМ
НЕРВНО-ПСИХИЧЕСКОГО РАЗВИТИЯ
Эти нарушения метаболизма органических

В 1967 году (Budd M. А. et al.) впервые была уточнена
В 1967 году (Budd M. А. et al.) впервые была уточнена

Первичные симптомы:
- респираторный и нейродистресс-синдромы,
- припадки,
- рвота, отказ от еды,
Первичные симптомы: - респираторный и нейродистресс-синдромы, - припадки, - рвота, отказ от еды,

Вторичные нарушения:
- отставание психического и моторного развития,
-пирамидная симптоматика,
-расстройства
Вторичные нарушения: - отставание психического и моторного развития, -пирамидная симптоматика, -расстройства

Лечение
ограничение белка;
высокие дозах витаминов;
дополнительное введение Л-карнитина и глицина.
Лечение
ограничение белка;
высокие дозах витаминов;
дополнительное введение Л-карнитина и глицина.

Галактоземия
Описана в 1908 году,
однако дефект обмена,
ее обуславливающий,
был открыт
Галактоземия
Описана в 1908 году,
однако дефект обмена,
ее обуславливающий,
был открыт

Патогенез
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар)
Патогенез
Дефицит фермента галактозо-1-фосфат-уридил-трансферазы (Г-1 -ФУТФ). В результате галактоза (молочный сахар)

Клиника
Проявляется вскоре после рождения у ребенка:
отказом от пищи, поносом, рвотой, непереносимостью
Клиника
Проявляется вскоре после рождения у ребенка:
отказом от пищи, поносом, рвотой, непереносимостью

Лечение
Безмолочная диета
Лечение
Безмолочная диета

МУКОПОЛИСАХАРИДО3 1 Н (СИНДРОМ ГУРЛЕРА)
Описан G. Gurler в 1919 году.
Встречается
МУКОПОЛИСАХАРИДО3 1 Н (СИНДРОМ ГУРЛЕРА)
Описан G. Gurler в 1919 году.
Встречается

Клиника
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова, выдающиеся
Клиника
Проявляется на первом году жизни.
Внешний вид больных — увеличенная голова, выдающиеся

Умственная отсталость заметна уже в раннем возрасте.
В последующем интеллектуальный дефект
Умственная отсталость заметна уже в раннем возрасте. В последующем интеллектуальный дефект

Патогенез
Отложение мукополисахаридов в соединительной ткани печени, селезенки и других тканях. Накопление
Патогенез
Отложение мукополисахаридов в соединительной ткани печени, селезенки и других тканях. Накопление

Болезнь Гирке
Нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях
Болезнь Гирке
Нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях

Альбинизм
При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене)
Альбинизм
При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене)

Алкаптонурия
Заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты —
Алкаптонурия
Заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты —

Гиперхолестеринемия
Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном
Гиперхолестеринемия
Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном
 Пилотный проект «Прямые выплаты» с 1 января 2019 года. Часть 2
Пилотный проект «Прямые выплаты» с 1 января 2019 года. Часть 2 Реклама, стимулирование сбыта
Реклама, стимулирование сбыта Презентация на тему "Методы обучения" - скачать презентации по Педагогике
Презентация на тему "Методы обучения" - скачать презентации по Педагогике Телевидение и передача видеосигналов в ТКС
Телевидение и передача видеосигналов в ТКС Право собственности
Право собственности ИСТОКИ: СЕМЬ ЧУДЕС РОССИИ Цель урока: введение в курс предмета «Истоки» Задачи: 1. знакомство с семью выдающимися памятниками отеч
ИСТОКИ: СЕМЬ ЧУДЕС РОССИИ Цель урока: введение в курс предмета «Истоки» Задачи: 1. знакомство с семью выдающимися памятниками отеч Предмет, метод и теоретические основы методов линейного программирования
Предмет, метод и теоретические основы методов линейного программирования Изображение колеса и радиуса
Изображение колеса и радиуса  Одноэтажные промышленные здания
Одноэтажные промышленные здания History of computers
History of computers Функция - презентация по Алгебре
Функция - презентация по Алгебре «Папа Карло». Рекламное агентство
«Папа Карло». Рекламное агентство Дизайны одноэтажных домов
Дизайны одноэтажных домов Проектирование базы данных (фирма –посредник)
Проектирование базы данных (фирма –посредник) Выборки
Выборки Новообразования кожи
Новообразования кожи  Ключи к здоровью позвоночника и суставов. Тренинг
Ключи к здоровью позвоночника и суставов. Тренинг Презентация Структура предприятия
Презентация Структура предприятия ХиТРРЭ.pptx
ХиТРРЭ.pptx Использование сайтов при подготовке к ЕГЭ
Использование сайтов при подготовке к ЕГЭ Design Patterns. Проверенные решения
Design Patterns. Проверенные решения Соленое тесто
Соленое тесто Linguistic Anthropology. Week 2
Linguistic Anthropology. Week 2 Презентация Анализ состава, структуры и динамики таможенных платежей на примере Центральной акцизной таможни
Презентация Анализ состава, структуры и динамики таможенных платежей на примере Центральной акцизной таможни Проект по биологии
Проект по биологии  Лекция 2. Основы разработки Web-приложений
Лекция 2. Основы разработки Web-приложений Методы экспертных оценок
Методы экспертных оценок Презентация "Бизнес-план" - скачать презентации по Экономике
Презентация "Бизнес-план" - скачать презентации по Экономике