- Генетика 9

Содержание
- 2. В лекции рассматриваются следующие вопросы: 1. Характеристика наследственных болезней человека 2. Хромосомные болезни: генетические основы их
- 3. КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ ЧЕЛОВЕКА 1. Хромосомные болезни Причина – изменение количества или структуры хромосом 2. Генные
- 4. Мультифакториальные болезни определяются полигенами , патологические проявления которых осуществляются во взаимодействии с факторами окружающей среды. При
- 5. Центромера - компактный участок хромосомы, к которому при делении клетки прикрепляются волокна веретена деления. Теломера -
- 6. КЛАССИФИКАЦИЯ ХРОМОСОМ ПО РАЗМЕРУ И ПОЛОЖЕНИЮ ЦЕНТРОМЕРЫ
- 7. C
- 8. Хромосомный набор мужчины
- 9. Анеуплоидии возникают вследствие нарушения распределения хромосом по дочерним клеткам в мейозе или в первых дроблениях зиготы.
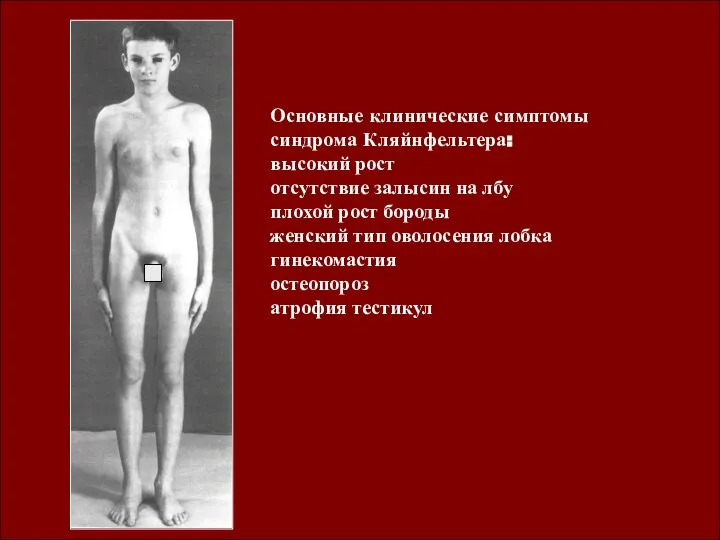
- 10. Синдром Клайнфельтера и синдром Шерешевского- Тернера как результат нарушения сперматогенеза
- 11. Синдром Клайнфельтера и синдром Шерешевского- Тернера как результат нарушения оогенеза
- 13. Геномным мутациям свойственны следующие особенности: недостаток генетического материала сопровождается более серьезными нарушениями, чем его избыток. Поэтому
- 14. Хромосомные мутации связанные со структурными перестройками хромосом подразделяют на две группы: а) мутации, которые приводят к
- 15. Дети с синдромом Дауна. А. Европеоид. Б.Негр. В. Представитель азиатской расы. Признаки синдрома Дауна более заметны
- 16. Дети разного возраста с характерными чертами синдрома Дауна: Круглое, лунообразное лицо , макроглоссия и открытый рот,
- 17. Девочкая с синдромом Дауна в возрасте 1 года и 7 лет
- 18. Задержка роста Умственная отсталость Плоский затылок Много петель на кончиках пальцев Одностороннее или двустороннее отсутствие ребра
- 19. Зависимость частоты рождения детей с синдромом Дауна от возраста матери
- 20. матери Зависимость частоты рождения детей с хромосомными болезнями от возраста матери
- 21. Основные клинические симптомы трисомии по 18 паре хромосом. Синдром Эдвардса (47, XY +18) Открытые швы черепа,
- 22. Основные клинические симптомы трисомии по 13 паре хромосом. Синдром Патау (47, XY +13) Микрофтальм, узкие глазные
- 23. Синдром Патау (47, XY +13)
- 26. Синдром Шерешевского-Тернера ( 45, ХО) Шейные крыловидные складки, широко расположенные и недоразвитые соски молочных желез ,
- 27. Нарушения, связанные с различными типами анеуплоидии у человека
- 28. Структура хромосомных аномалий ( все аномалии приняты за 100 % )
- 29. Распределение генов участвующих в основных процессах жизнедеятельности клетки ( в %): синтез белков и РНК- 22;
- 30. Время проявления болезни в онтогенезе
- 31. Болезнь Альцгеймера Аллель гена аполипопротеина Е Гены пресинилина Гены бета – амилоида 21 хромосоме.
- 32. Частота новорожденных с генными болезнями ( в %)
- 33. В зависимости от характера мутации и подверженности ее популяционным сдвигам моногенные болезни подразделяют на две группы:
- 34. Синдром Марфана 15q21 ( ген фибриллина ) Высокий рост, длинные тонкие « паучьи пальцы , вывих
- 35. «мертвенно-бледное, как будто вылепленное из воска лицо, глубоко запавшие глаза, худоба, угловатые движения и, самое главное,
- 36. Ахондроплазия
- 37. Нейрофиброматоз 1-го типа, 17 q, ген- 350000 п.о.
- 38. Хорея Хантингтона Ген расположен в 4 хромосоме (IT-15 4p16.3) . Болезнь связана с нарастанием числа повторов
- 39. Синдром фрагильной (ломкой) Х-хромосомы характеризуется умственной отсталостью и рядом физических пороков развития (большие яички, большие оттопыренные
- 40. Гемофилия А 10 q 186000 п.о. 26 экзонов, ген кодирует VIII фактор свертывания крови,. Кровотечения ,
- 41. Миодистрофия Дюшенна Х-сцепленный, Хр 2000000 п.о.ген кодирует белок дистрофин. Длина маричной днк 16000 по
- 42. Миодистрофия Дюшенна Х-сцепленный, рХ 2000000 п.о.
- 43. Синдром Леша —Найхана —заболевание, сцепленное с Х-хромосомой. Недостаточность гипоксантингуанинфосфорибозил- трансферазы и накопление мочевой кислоты. У больных
- 44. Частота аутосомно-рецессивных заболеваний ( на 10 тыс.)
- 45. Поражение легких при мукововисцидозе Ген расположен в 7 хромосоме ( 7q31-32) , размер составляет 250000 п.о
- 46. Трансмембранный белок – переносчик ионов хлора
- 47. Cерповидно-клеточная анемия Ген расположен в 11 хромосоме, «миссенс – мутация» замена в 6 положении бетта –
- 48. Для наследственных энзимопатий характерны следующие общие признаки: а) болезнь проявляются только у гомозигот, у гетерозигот присутствие
- 49. Больной с фенилкетонурией Слабая пигментация кожи, радужной оболочки глаз умеренная степень олигофрении (12q22-24)
- 50. ГАЛАКТОЗЕМИЯ - наследственное заболевание, в основе которого лежит метаболический блок на пути преобразования галактозы в глюкозу.
- 52. Синдром Прадера-Вилли Синдром Энгельмана Механизм возникновения синдромов Прадера Вилли и Энгельмана.
- 53. При обоих нарушениях генетическая причина одна и та же: делеция особого участка хромосомы 15 (15q11 –q13),
- 54. Мультифакториальное наследование в сочетании с пороговым эффектом Подверженность заболеванию в общей популяции имеет нормальное распределение ;
- 55. Гипотетические кривые распределения лиц с мультифакториальной предрасположенностью (популяция и родственники больных I степени родства По оси
- 56. Примечание: ИБС— ишемическая болезнь сердца; Р — ревматизм; СД — сахарный диабет; ЯБ — язвенная болезнь;
- 57. Показатели наследуемости на основании данных близнецовых исследований
- 58. Влияние генетических факторов на заболеваемость диабетом аборигенов Австралии и эскимосов Гренландии
- 59. Влияние генетических и средовых факторов на заболеваемость диабетом индусов.
- 60. Прото-онкоген Транспозиция Амплификация Мутация Нормальные белки, стимулирующие рост и деление клетки в избытке Нормальные белки, стимулирующие
- 62. Скачать презентацию

В лекции рассматриваются следующие вопросы:
1. Характеристика наследственных болезней человека
2. Хромосомные болезни:
В лекции рассматриваются следующие вопросы:
1. Характеристика наследственных болезней человека
2. Хромосомные болезни:

КЛАССИФИКАЦИЯ
НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ ЧЕЛОВЕКА
1. Хромосомные болезни
Причина – изменение количества
КЛАССИФИКАЦИЯ
НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ ЧЕЛОВЕКА
1. Хромосомные болезни
Причина – изменение количества

Мультифакториальные болезни определяются полигенами , патологические проявления которых осуществляются во
Мультифакториальные болезни определяются полигенами , патологические проявления которых осуществляются во
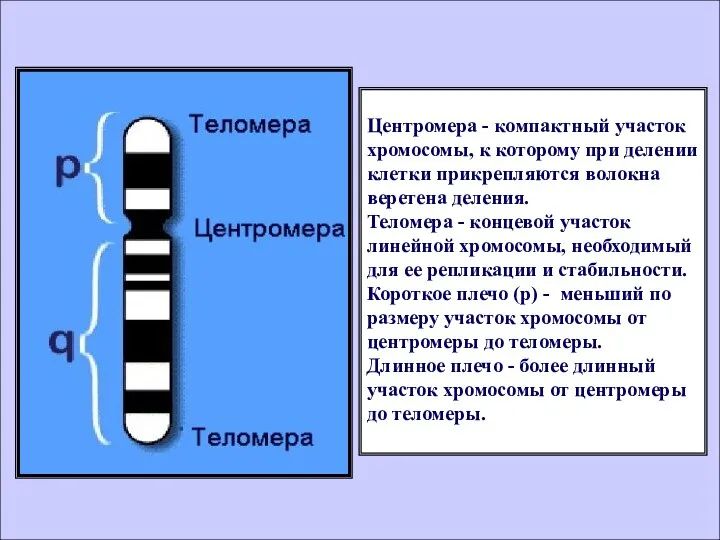
Центромера - компактный участок хромосомы, к которому при делении клетки прикрепляются

КЛАССИФИКАЦИЯ ХРОМОСОМ ПО РАЗМЕРУ И ПОЛОЖЕНИЮ ЦЕНТРОМЕРЫ
КЛАССИФИКАЦИЯ ХРОМОСОМ ПО РАЗМЕРУ И ПОЛОЖЕНИЮ ЦЕНТРОМЕРЫ
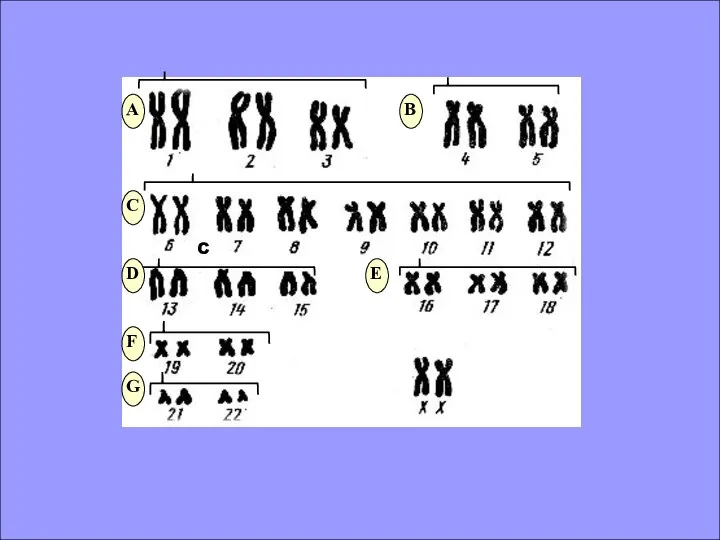
C
C
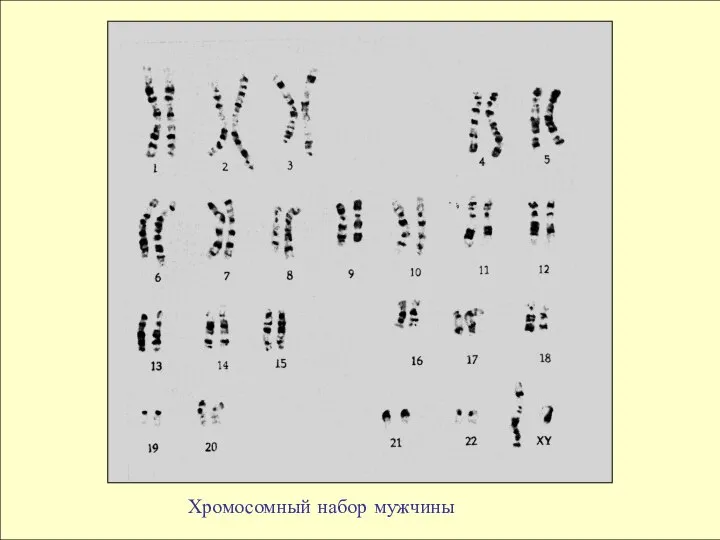
Хромосомный набор мужчины
Хромосомный набор мужчины

Анеуплоидии возникают вследствие нарушения распределения хромосом по дочерним клеткам в мейозе
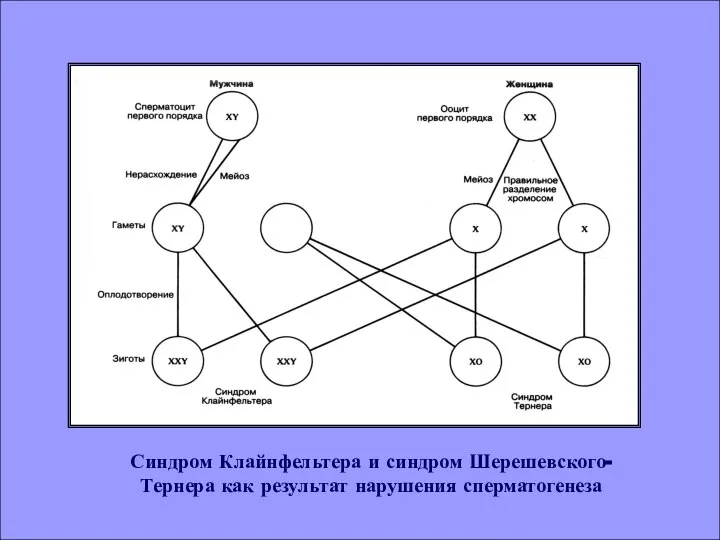
Синдром Клайнфельтера и синдром Шерешевского- Тернера как результат нарушения сперматогенеза
Синдром Клайнфельтера и синдром Шерешевского- Тернера как результат нарушения сперматогенеза
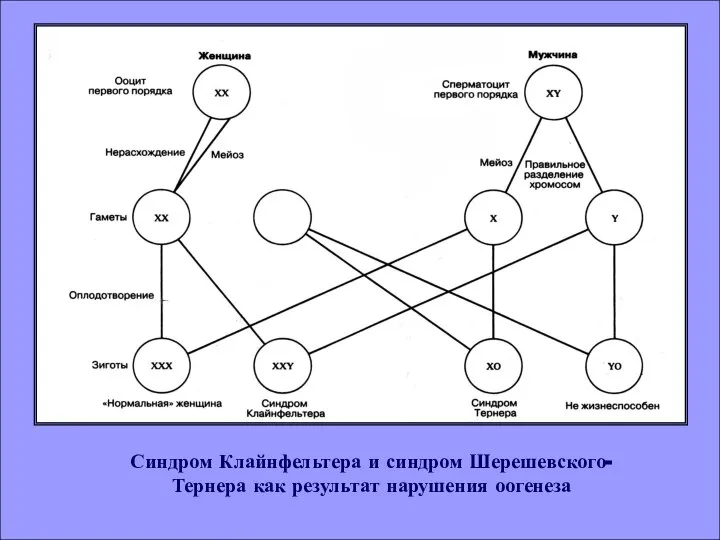
Синдром Клайнфельтера и синдром Шерешевского- Тернера как результат нарушения оогенеза
Синдром Клайнфельтера и синдром Шерешевского- Тернера как результат нарушения оогенеза
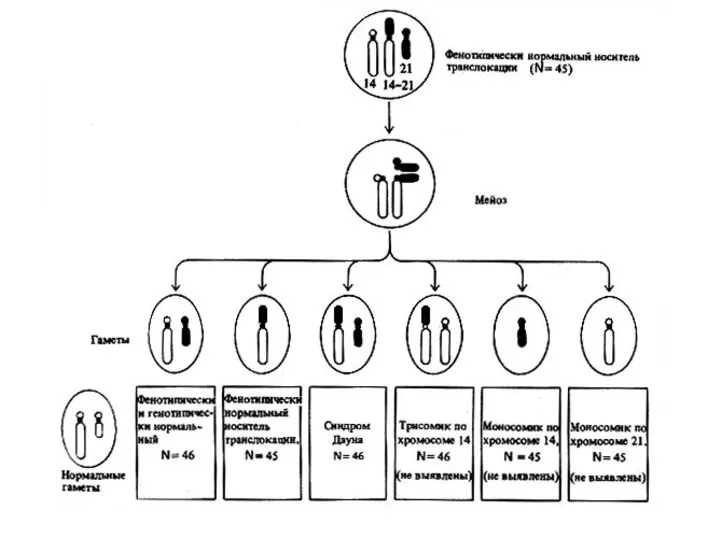

Геномным мутациям свойственны следующие особенности: недостаток генетического материала сопровождается более серьезными

Хромосомные мутации связанные со структурными перестройками хромосом подразделяют на две группы:
а)
а)
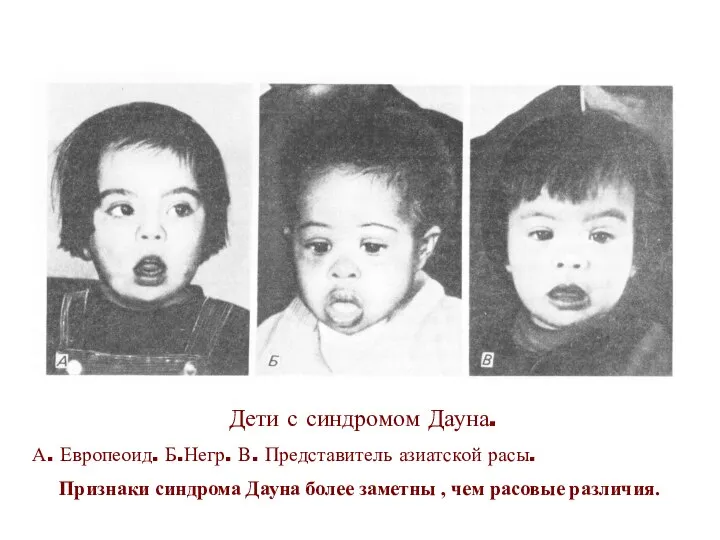
Дети с синдромом Дауна.
А. Европеоид. Б.Негр. В. Представитель азиатской расы.
Дети с синдромом Дауна.
А. Европеоид. Б.Негр. В. Представитель азиатской расы.
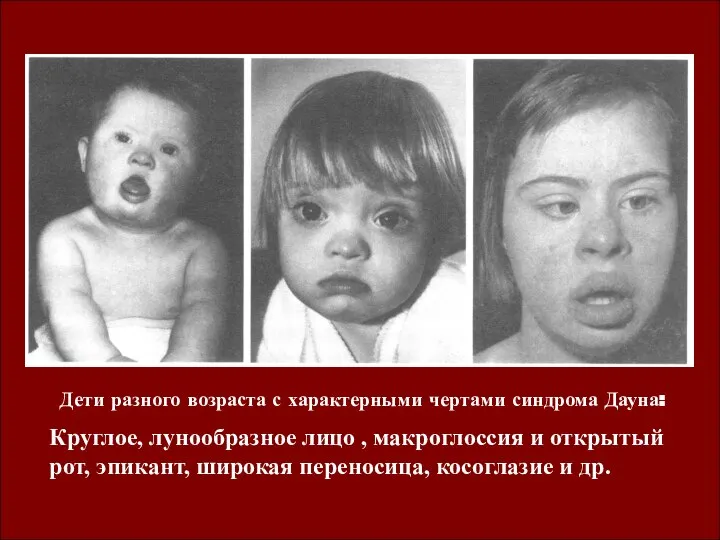
Дети разного возраста с характерными чертами синдрома Дауна:
Круглое, лунообразное лицо ,
Дети разного возраста с характерными чертами синдрома Дауна:
Круглое, лунообразное лицо ,
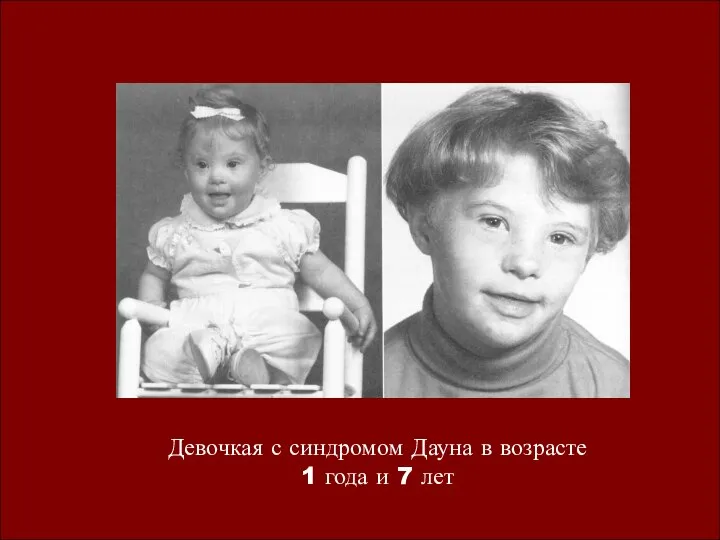
Девочкая с синдромом Дауна в возрасте
1 года и 7 лет
Девочкая с синдромом Дауна в возрасте
1 года и 7 лет
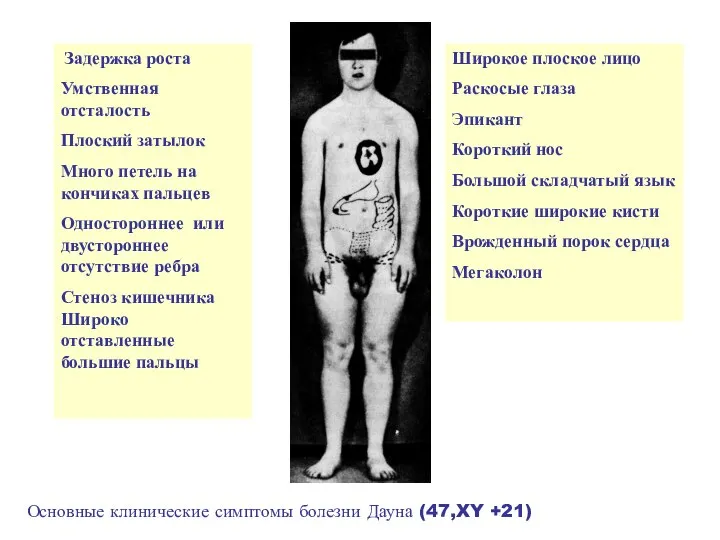
Задержка роста
Умственная отсталость
Плоский затылок
Много петель на кончиках пальцев
Одностороннее
Задержка роста
Умственная отсталость
Плоский затылок
Много петель на кончиках пальцев
Одностороннее
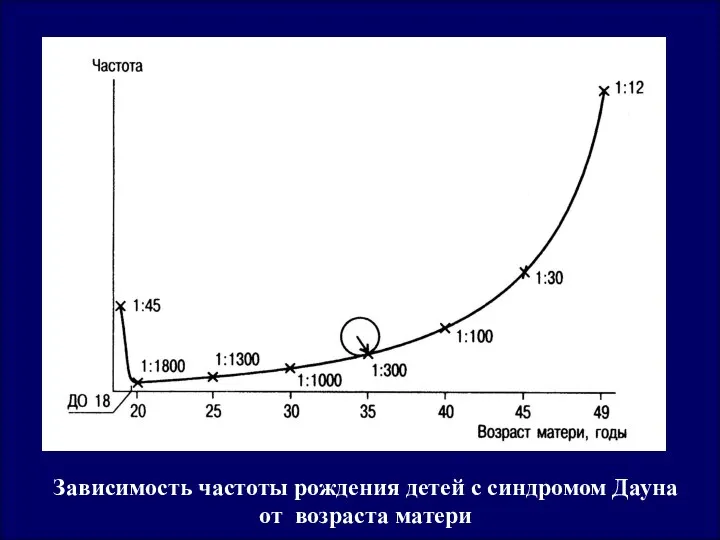
Зависимость частоты рождения детей с синдромом Дауна от возраста матери
Зависимость частоты рождения детей с синдромом Дауна от возраста матери
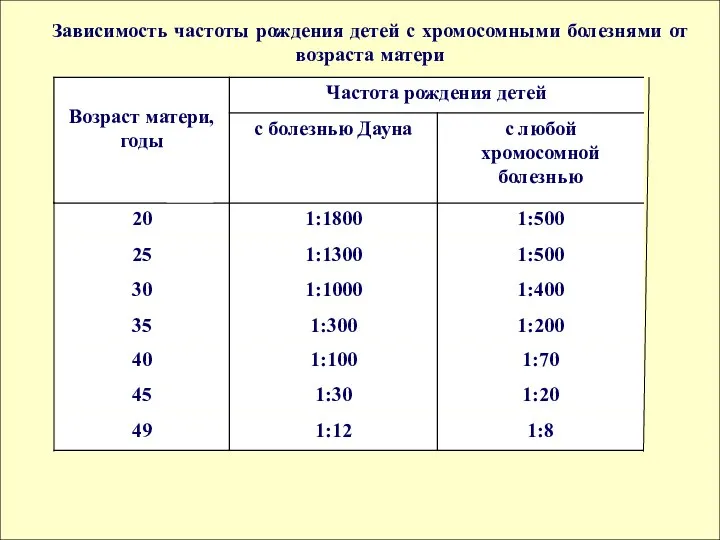
матери
Зависимость частоты рождения детей с хромосомными болезнями от возраста матери
матери
Зависимость частоты рождения детей с хромосомными болезнями от возраста матери

Основные клинические симптомы трисомии по 18 паре хромосом. Синдром Эдвардса (47,
Основные клинические симптомы трисомии по 18 паре хромосом. Синдром Эдвардса (47,
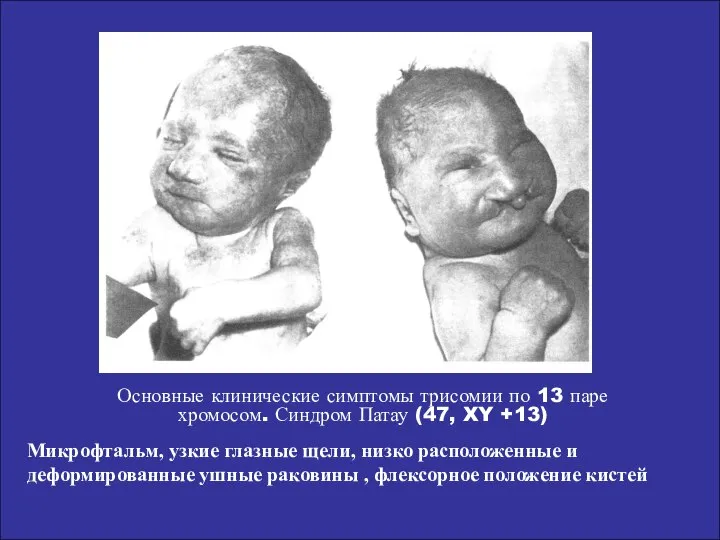
Основные клинические симптомы трисомии по 13 паре
хромосом. Синдром Патау (47, XY
Основные клинические симптомы трисомии по 13 паре
хромосом. Синдром Патау (47, XY
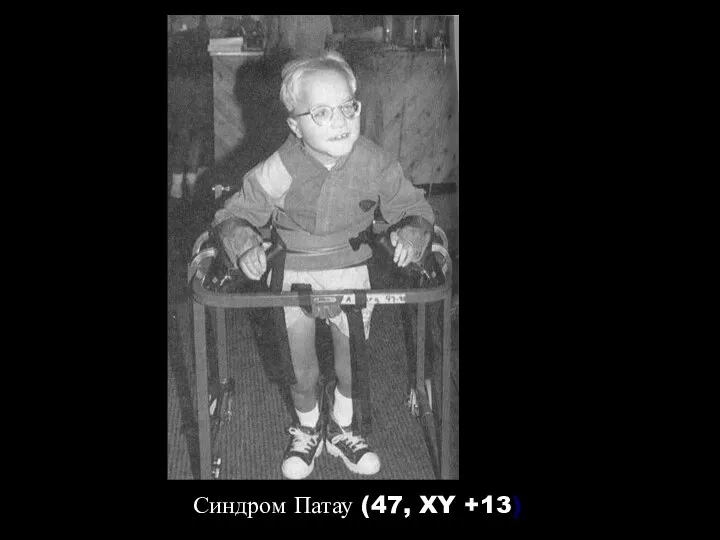
Синдром Патау (47, XY +13)
Синдром Патау (47, XY +13)
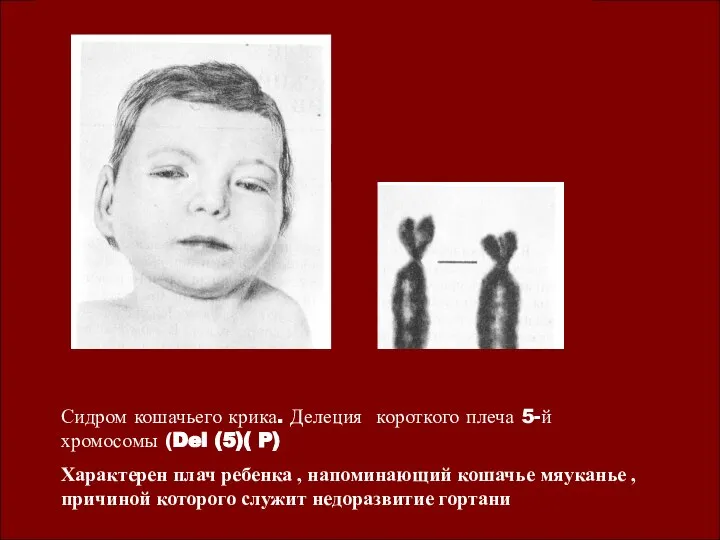
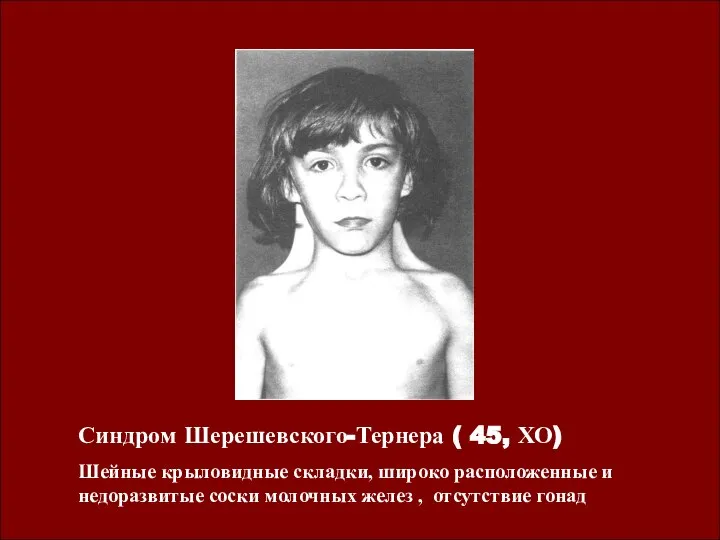
Синдром Шерешевского-Тернера ( 45, ХО)
Шейные крыловидные складки, широко расположенные и недоразвитые
Синдром Шерешевского-Тернера ( 45, ХО)
Шейные крыловидные складки, широко расположенные и недоразвитые
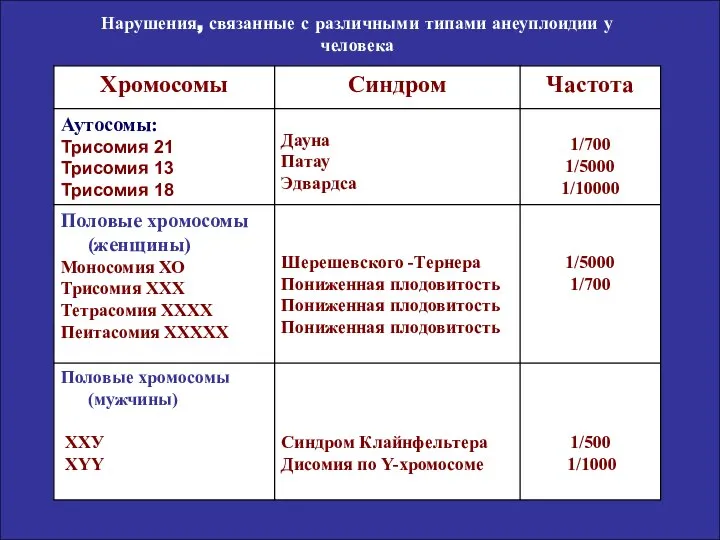
Нарушения, связанные с различными типами анеуплоидии у человека
Нарушения, связанные с различными типами анеуплоидии у человека

Структура хромосомных аномалий
( все аномалии приняты за 100 % )
Структура хромосомных аномалий
( все аномалии приняты за 100 % )

Распределение генов участвующих в основных процессах жизнедеятельности клетки ( в %):
синтез
Распределение генов участвующих в основных процессах жизнедеятельности клетки ( в %):
синтез
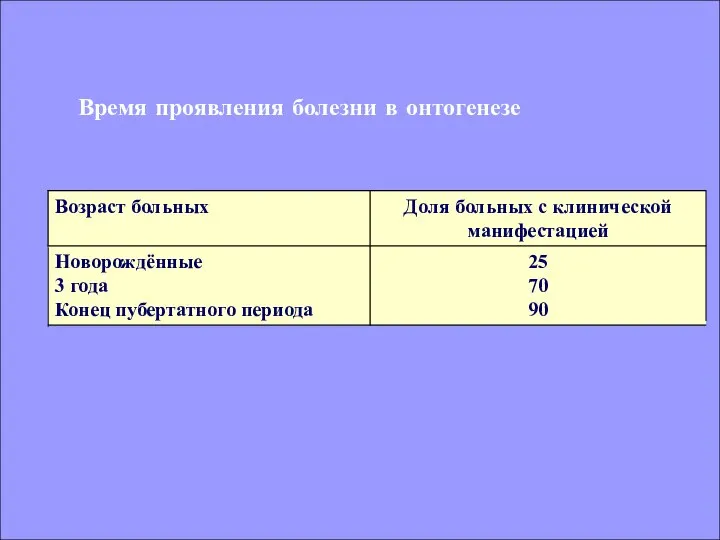
Время проявления болезни в онтогенезе
Время проявления болезни в онтогенезе

Болезнь Альцгеймера
Аллель гена аполипопротеина Е
Гены пресинилина
Гены бета – амилоида 21 хромосоме.
Болезнь Альцгеймера
Аллель гена аполипопротеина Е
Гены пресинилина
Гены бета – амилоида 21 хромосоме.

Частота новорожденных с генными болезнями ( в %)
Частота новорожденных с генными болезнями ( в %)

В зависимости от характера мутации и подверженности ее популяционным сдвигам
В зависимости от характера мутации и подверженности ее популяционным сдвигам

Синдром Марфана
15q21 ( ген фибриллина )
Высокий рост, длинные тонкие «
Синдром Марфана
15q21 ( ген фибриллина )
Высокий рост, длинные тонкие «

«мертвенно-бледное, как будто вылепленное из воска лицо, глубоко запавшие глаза, худоба,
«мертвенно-бледное, как будто вылепленное из воска лицо, глубоко запавшие глаза, худоба,

Ахондроплазия
Ахондроплазия
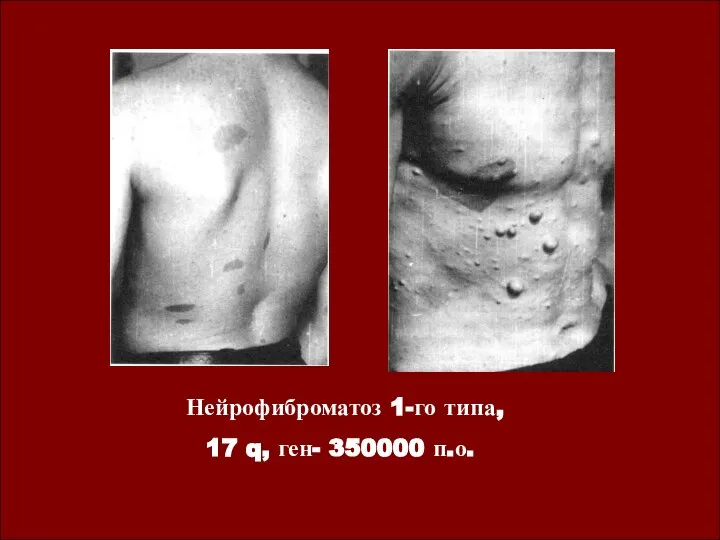
Нейрофиброматоз 1-го типа,
17 q, ген- 350000 п.о.
Нейрофиброматоз 1-го типа,
17 q, ген- 350000 п.о.
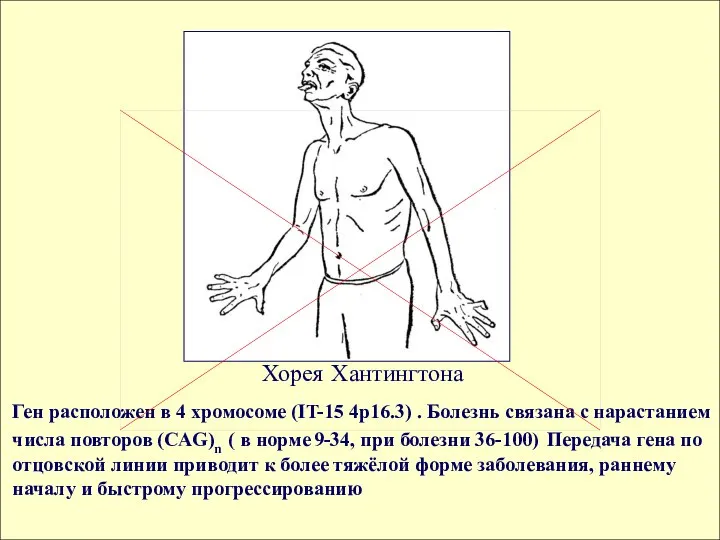
Хорея Хантингтона
Ген расположен в 4 хромосоме (IT-15 4p16.3) . Болезнь
Хорея Хантингтона
Ген расположен в 4 хромосоме (IT-15 4p16.3) . Болезнь

Синдром фрагильной (ломкой) Х-хромосомы характеризуется умственной отсталостью и рядом физических пороков
Синдром фрагильной (ломкой) Х-хромосомы характеризуется умственной отсталостью и рядом физических пороков
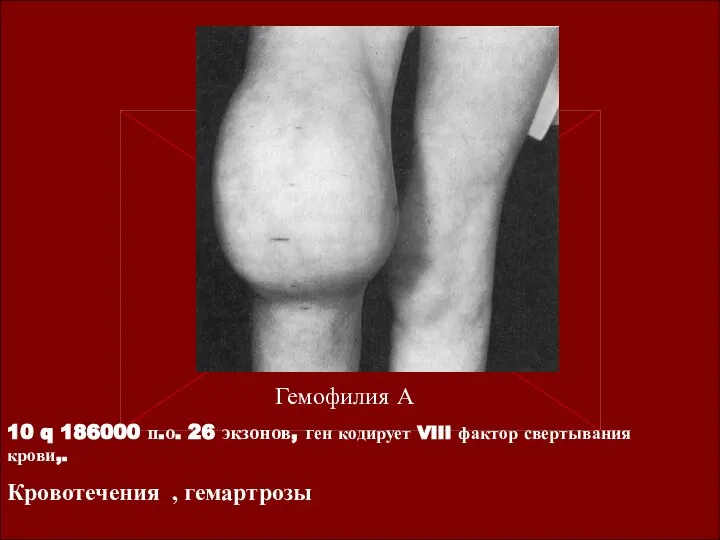
Гемофилия А
10 q 186000 п.о. 26 экзонов, ген кодирует VIII фактор
Гемофилия А
10 q 186000 п.о. 26 экзонов, ген кодирует VIII фактор
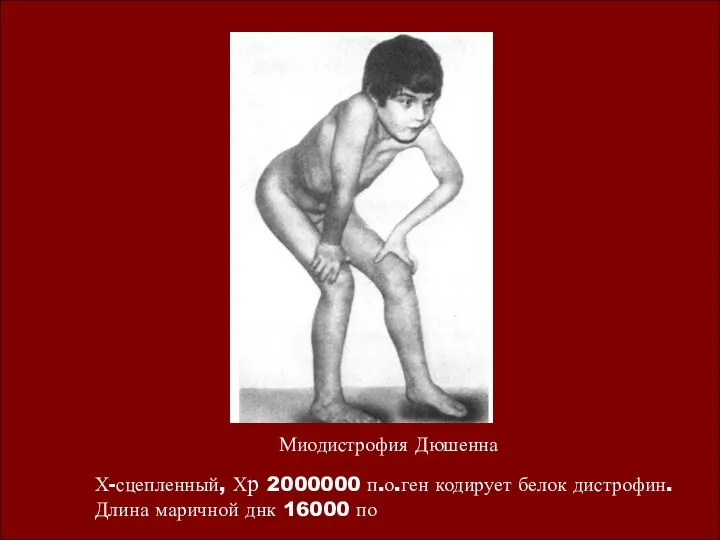
Миодистрофия Дюшенна
Х-сцепленный, Хр 2000000 п.о.ген кодирует белок дистрофин. Длина маричной днк
Миодистрофия Дюшенна
Х-сцепленный, Хр 2000000 п.о.ген кодирует белок дистрофин. Длина маричной днк
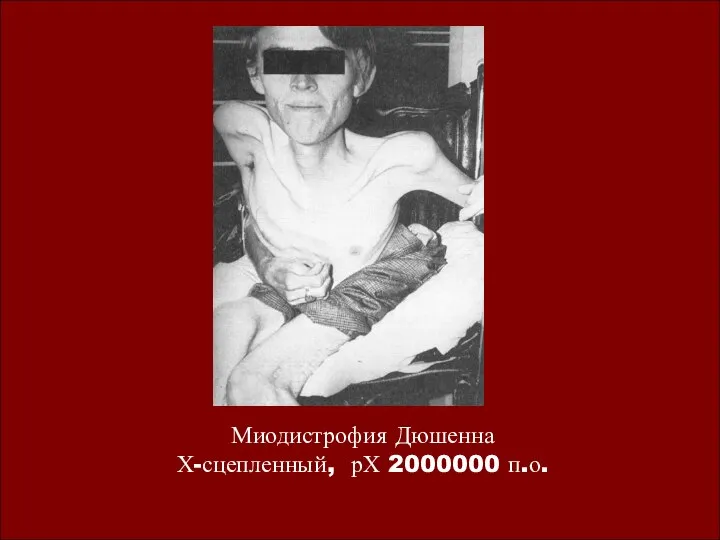
Миодистрофия Дюшенна
Х-сцепленный, рХ 2000000 п.о.
Миодистрофия Дюшенна
Х-сцепленный, рХ 2000000 п.о.

Синдром Леша —Найхана —заболевание, сцепленное с Х-хромосомой.
Недостаточность гипоксантингуанинфосфорибозил-
трансферазы и накопление
Синдром Леша —Найхана —заболевание, сцепленное с Х-хромосомой.
Недостаточность гипоксантингуанинфосфорибозил-
трансферазы и накопление
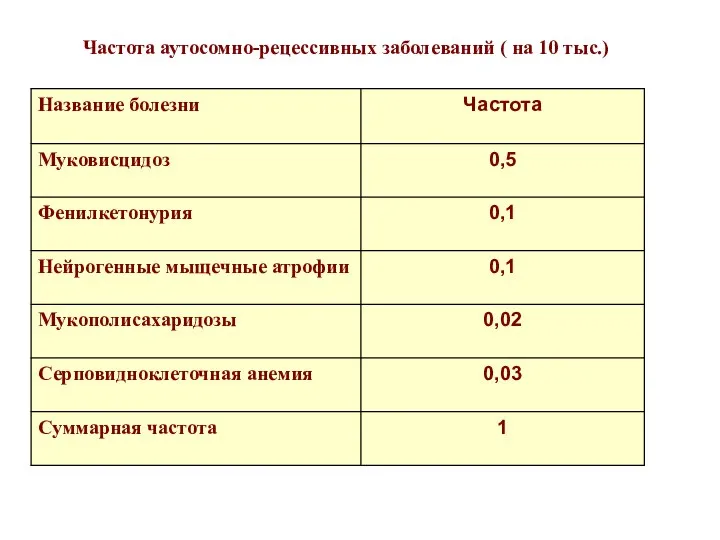
Частота аутосомно-рецессивных заболеваний ( на 10 тыс.)
Частота аутосомно-рецессивных заболеваний ( на 10 тыс.)
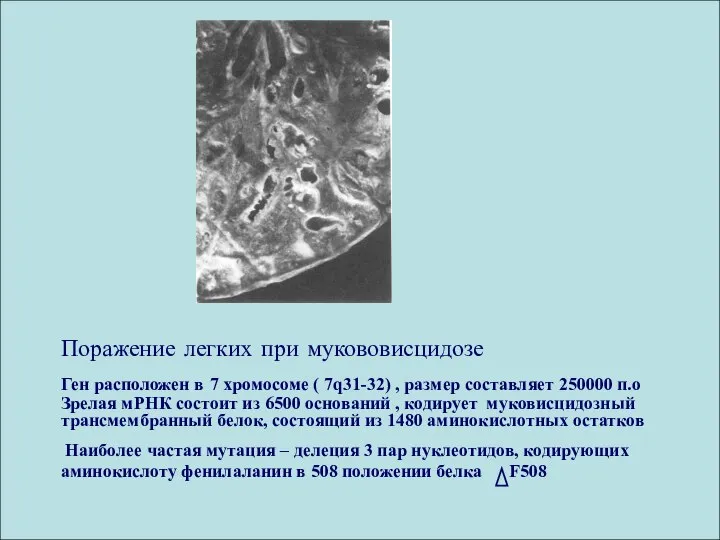
Поражение легких при мукововисцидозе
Ген расположен в 7 хромосоме ( 7q31-32) ,
Поражение легких при мукововисцидозе
Ген расположен в 7 хромосоме ( 7q31-32) ,
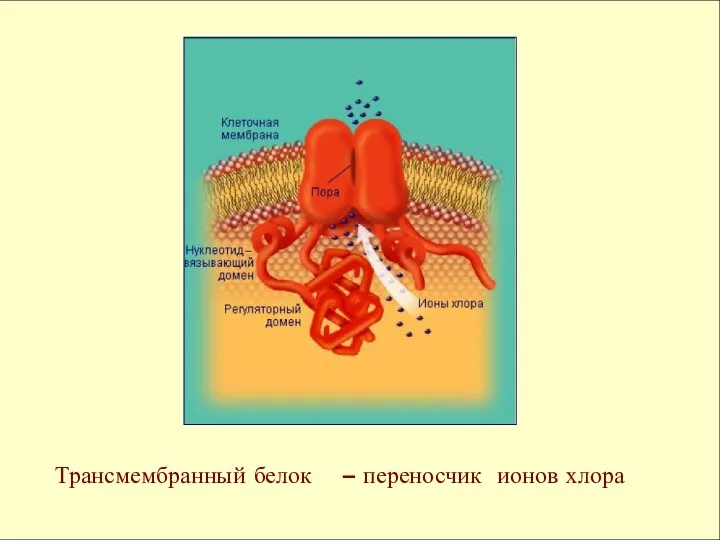
Трансмембранный белок – переносчик ионов хлора
Трансмембранный белок – переносчик ионов хлора
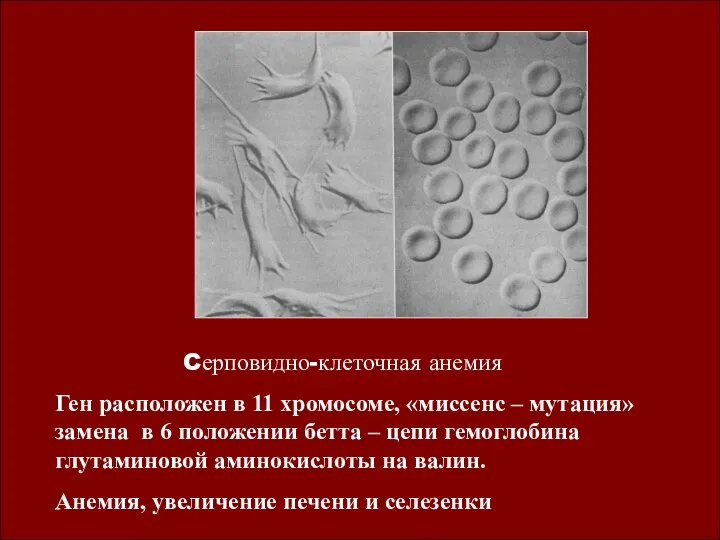
Cерповидно-клеточная анемия
Ген расположен в 11 хромосоме, «миссенс – мутация» замена
Cерповидно-клеточная анемия
Ген расположен в 11 хромосоме, «миссенс – мутация» замена

Для наследственных энзимопатий характерны следующие общие признаки:
а) болезнь проявляются только
Для наследственных энзимопатий характерны следующие общие признаки:
а) болезнь проявляются только

Больной с фенилкетонурией
Слабая пигментация кожи, радужной оболочки глаз умеренная
Больной с фенилкетонурией
Слабая пигментация кожи, радужной оболочки глаз умеренная

ГАЛАКТОЗЕМИЯ - наследственное заболевание, в основе которого лежит метаболический блок на
ГАЛАКТОЗЕМИЯ - наследственное заболевание, в основе которого лежит метаболический блок на
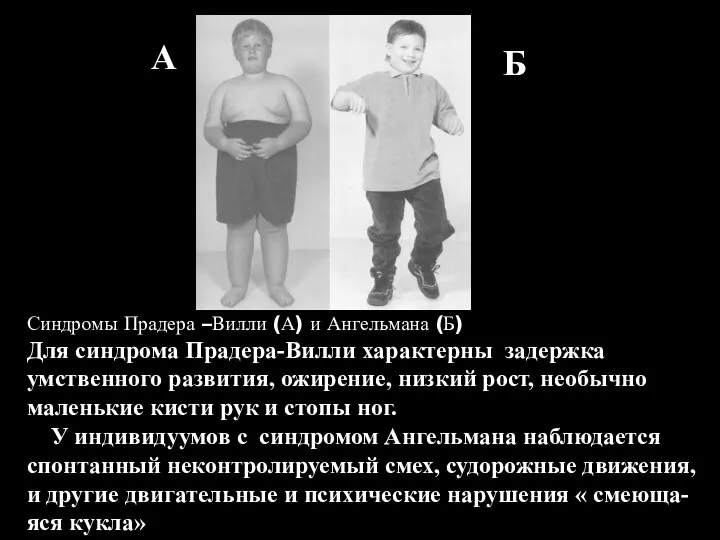
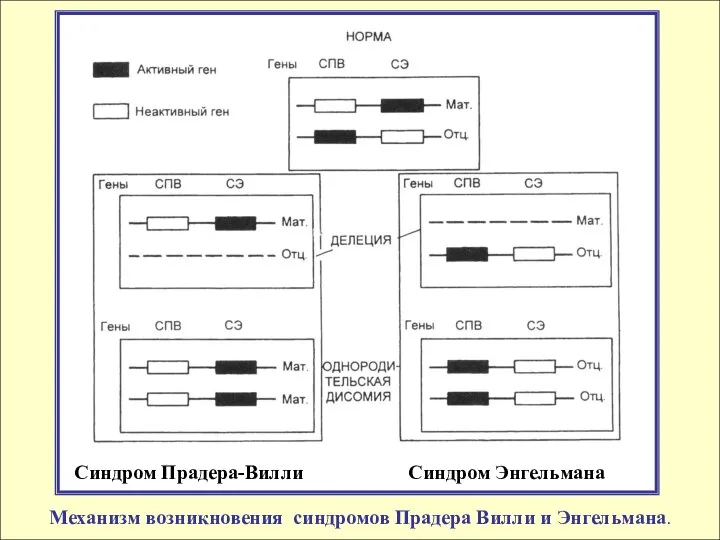
Синдром Прадера-Вилли Синдром Энгельмана
Механизм возникновения синдромов Прадера Вилли и
Синдром Прадера-Вилли Синдром Энгельмана
Механизм возникновения синдромов Прадера Вилли и

При обоих нарушениях генетическая причина одна и та же: делеция особого
При обоих нарушениях генетическая причина одна и та же: делеция особого
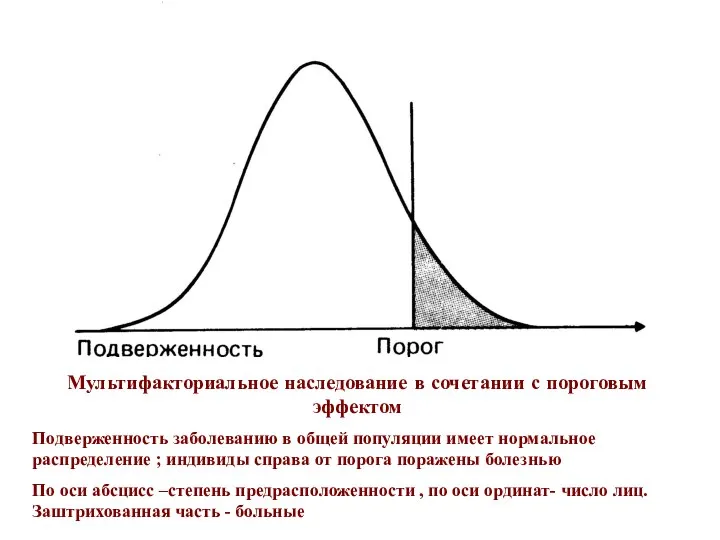
Мультифакториальное наследование в сочетании с пороговым эффектом
Подверженность заболеванию в общей
Мультифакториальное наследование в сочетании с пороговым эффектом
Подверженность заболеванию в общей
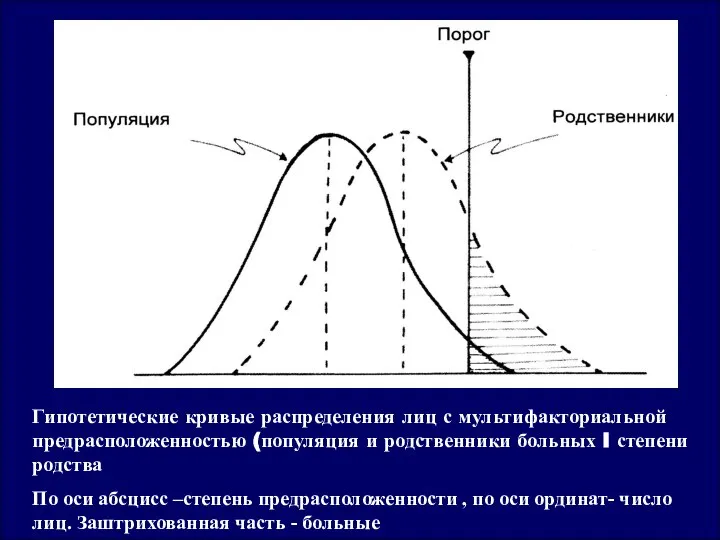
Гипотетические кривые распределения лиц с мультифакториальной предрасположенностью (популяция и родственники больных
Гипотетические кривые распределения лиц с мультифакториальной предрасположенностью (популяция и родственники больных
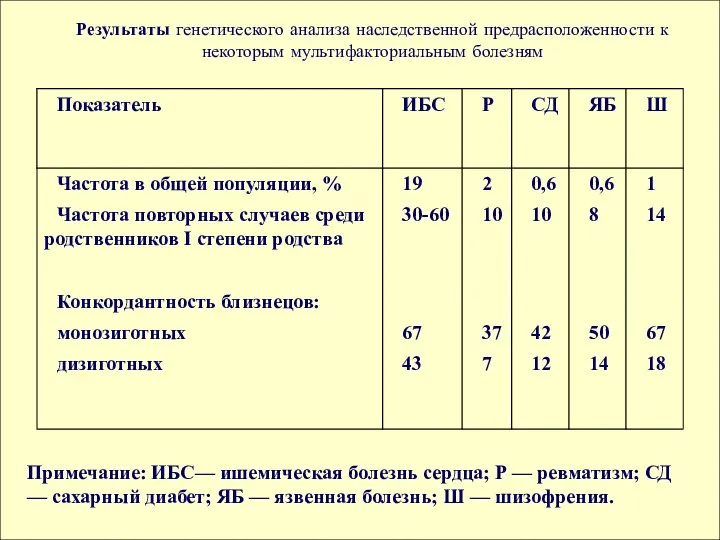
Примечание: ИБС— ишемическая болезнь сердца; Р — ревматизм; СД — сахарный
Примечание: ИБС— ишемическая болезнь сердца; Р — ревматизм; СД — сахарный
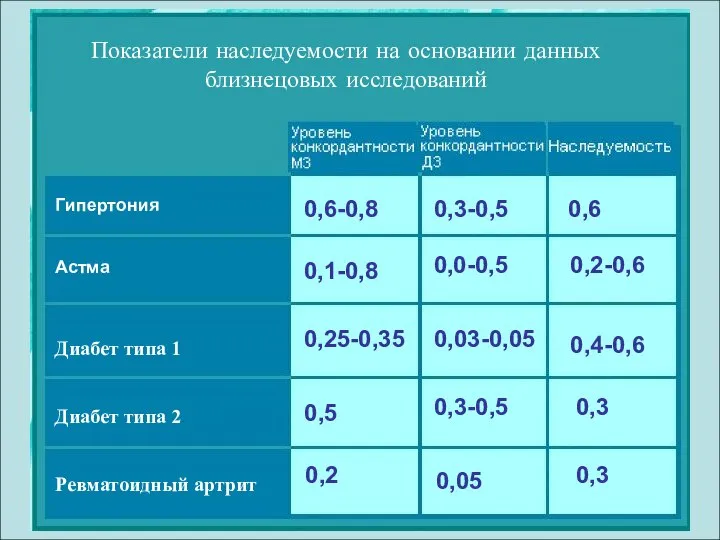
Показатели наследуемости на основании данных близнецовых исследований
Показатели наследуемости на основании данных близнецовых исследований

Влияние генетических факторов на заболеваемость диабетом аборигенов Австралии и эскимосов Гренландии
Влияние генетических факторов на заболеваемость диабетом аборигенов Австралии и эскимосов Гренландии
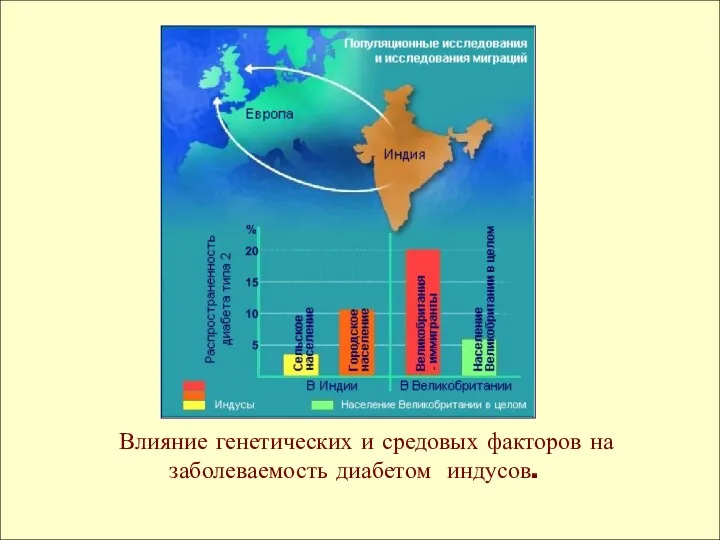
Влияние генетических и средовых факторов на заболеваемость диабетом индусов.
Влияние генетических и средовых факторов на заболеваемость диабетом индусов.
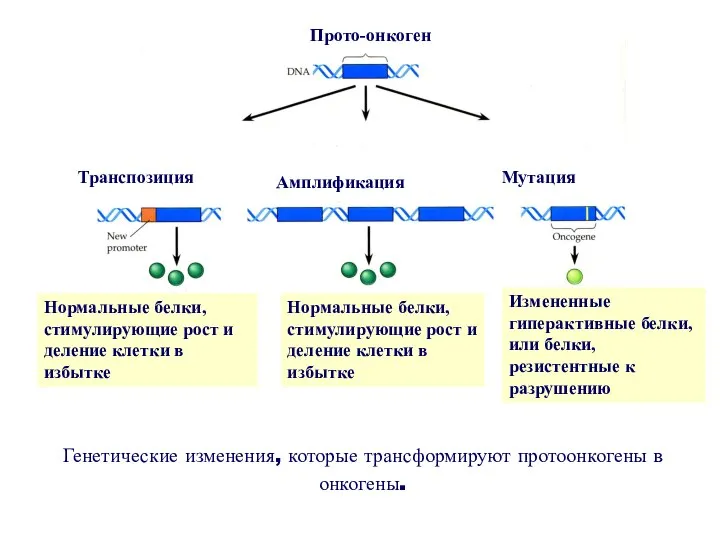
Прото-онкоген
Транспозиция
Амплификация
Мутация
Нормальные белки, стимулирующие рост и деление клетки в избытке
Нормальные
Прото-онкоген
Транспозиция
Амплификация
Мутация
Нормальные белки, стимулирующие рост и деление клетки в избытке
Нормальные
 Классы. ООП в Java. Конструкторы. Блоки инициализации
Классы. ООП в Java. Конструкторы. Блоки инициализации Дымковская игрушка
Дымковская игрушка Работа на радиостанциях КВ и УКВ диапазонов. Антенны военных радиостанций. (Тема 5.1)
Работа на радиостанциях КВ и УКВ диапазонов. Антенны военных радиостанций. (Тема 5.1) Електронна демократія та електронна держава
Електронна демократія та електронна держава Качественное сказуемое. Китайский язык
Качественное сказуемое. Китайский язык Vue JS The Progressive JavaScript Framework
Vue JS The Progressive JavaScript Framework 1 апреля - день шуток и смеха
1 апреля - день шуток и смеха Краснуха
Краснуха  Формулы приведения
Формулы приведения Презентация на тему Сыр
Презентация на тему Сыр Линия. Штрих
Линия. Штрих Красная шапочка - презентация для начальной школы_
Красная шапочка - презентация для начальной школы_ Презентация История Появление института президентства
Презентация История Появление института президентства  Обследование технического состояния строительных конструкций зданий и сооружений
Обследование технического состояния строительных конструкций зданий и сооружений Духовная жизнь 20-30 годов
Духовная жизнь 20-30 годов Программирование на языке Паскаль. Оператор выбора
Программирование на языке Паскаль. Оператор выбора Физическая культура людей молодого и зрелого возраста
Физическая культура людей молодого и зрелого возраста Sztos. Инструкция оплаты через терминал
Sztos. Инструкция оплаты через терминал Особенности празднования Нового года и Рождества в разных странах
Особенности празднования Нового года и Рождества в разных странах Medic Control Peel Линия средств для химического пилинга
Medic Control Peel Линия средств для химического пилинга Накопители на магнитных дисках
Накопители на магнитных дисках Презентация ГАТТ-1994
Презентация ГАТТ-1994 Функциональные схемы систем автоматизации технологических процессов
Функциональные схемы систем автоматизации технологических процессов Основные способы получения промежуточных температур
Основные способы получения промежуточных температур Гражданство Российской Федерации
Гражданство Российской Федерации Газовые Приборы
Газовые Приборы Кинематографическая журналистика. Аудиовизуальное повествование в интернете
Кинематографическая журналистика. Аудиовизуальное повествование в интернете Казахстано-саудовские отношения
Казахстано-саудовские отношения