- Квантовая химия молекул

Содержание
- 2. молекулярная динамика и метод Монте-Карло неэмпирическая квантовая химия полуэмпирическая квантовая химия квантовая статистическая механика молекулярная механика
- 3. Квантовая химия молекулы Энергия молекулы состоит из кинетических энергий каждого электрона и ядра и попарных энергий
- 4. Основное уравнение молекулярной квантовой химии - независящее от времени нерелятивистское уравнение Шредингера: Н Ψ ({r, R})
- 5. Приближение Борна-Оппенгеймера Электрон-ядерное взаимодействие Vэя значительно и пренебречь им нельзя. Hмол зависит от координат электронов и
- 6. 2) mэ/Mz ≤ 1/1836 и движение ядер много медленнее, чем электронов. Для большинства задач структурной химии
- 7. Молекулярная структура Ядерная конфигурация молекулы стабильна относительно малых колебаний ядер и характеризует молекулярную структуру. Топология ППЭ
- 8. 2) Молекулярная структура в пределах структурной области сохраняет одинаковую систему химических связей при разной геометрии. Если
- 9. Энергии вращательных барьеров (ккал/моль) После символа // указан базис, в котором была оптимизирована геометрия. Молекулярная структура
- 10. 2.3 Метод Хартри-Фока для молекул Зафиксировав ядерную конфигурацию, для анализа электронного поведения молекул достаточно рассматривать только
- 11. Полная ХФ энергия молекулы с замкнутыми оболочками (2.11) Замечания. 1) В минимизации энергии участвуют только занятые
- 12. Приближение МО ЛКАО. Уравнения Рутана Уравнения Хартри-Фока для молекул Численное решение МО электрона, который находится в
- 13. (2.15) (2.16) (2.17) (2.18) Еμ - одно из решений секулярного уравнения Удобно ввести матрицу зарядов- порядков
- 14. Электронная энергия молекулы с закрытыми оболочками в терминах введенных обозначений записывается в методе Рутана следующим образом:
- 15. Блок-схема вычислительного процесса решения уравнений Рутана
- 16. Блок-схема вычислительного процесса решения уравнений Рутана
- 17. Из-за наличия самосогласованного поля уравнения ХФ нелинейны: решения можно получить, лишь задав некоторый потенциал, обусловленный распределением
- 18. Многоэлектронная волновая функция и энергии состояний, получаемые с помощью метода Рутана (и с помощью метода Хартри-Фока
- 19. 4. Ограничения метода Хартри-Фока В ряде молекулярных задач, решаемых методом ХФ, проявляется так называемая "дилемма симметрии".
- 21. Скачать презентацию
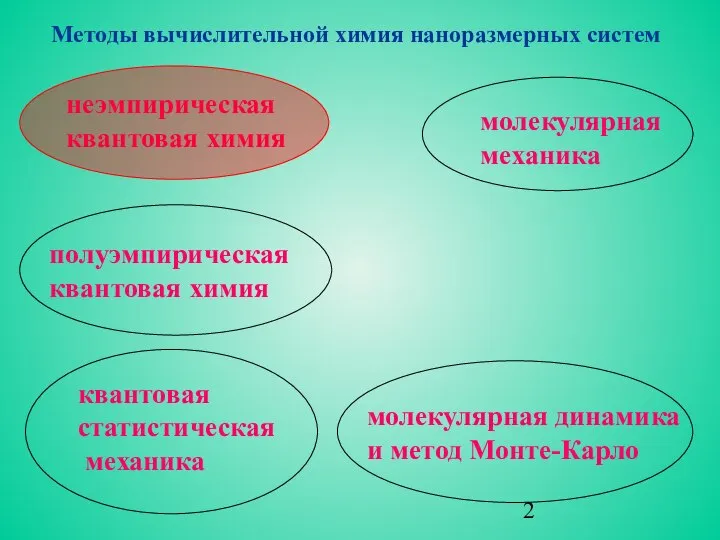
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия
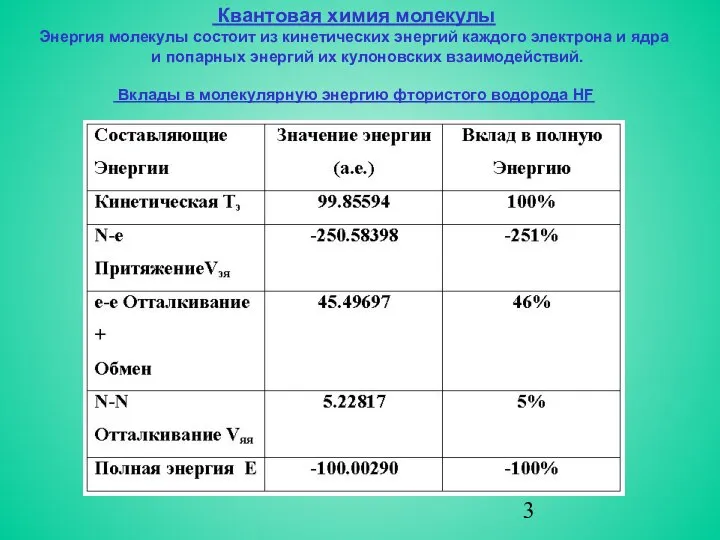
Квантовая химия молекулы
Энергия молекулы состоит из кинетических энергий каждого электрона
Квантовая химия молекулы
Энергия молекулы состоит из кинетических энергий каждого электрона

Основное уравнение молекулярной квантовой химии - независящее от времени нерелятивистское уравнение
Основное уравнение молекулярной квантовой химии - независящее от времени нерелятивистское уравнение

Приближение Борна-Оппенгеймера
Электрон-ядерное взаимодействие Vэя значительно и пренебречь им нельзя. Hмол зависит
Приближение Борна-Оппенгеймера
Электрон-ядерное взаимодействие Vэя значительно и пренебречь им нельзя. Hмол зависит
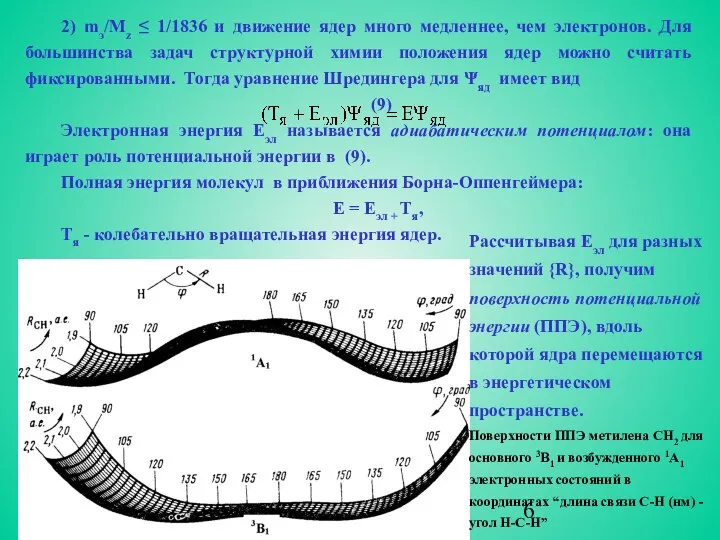
2) mэ/Mz ≤ 1/1836 и движение ядер много медленнее, чем электронов.
2) mэ/Mz ≤ 1/1836 и движение ядер много медленнее, чем электронов.

Молекулярная структура
Ядерная конфигурация молекулы стабильна относительно малых колебаний ядер и характеризует
Молекулярная структура
Ядерная конфигурация молекулы стабильна относительно малых колебаний ядер и характеризует
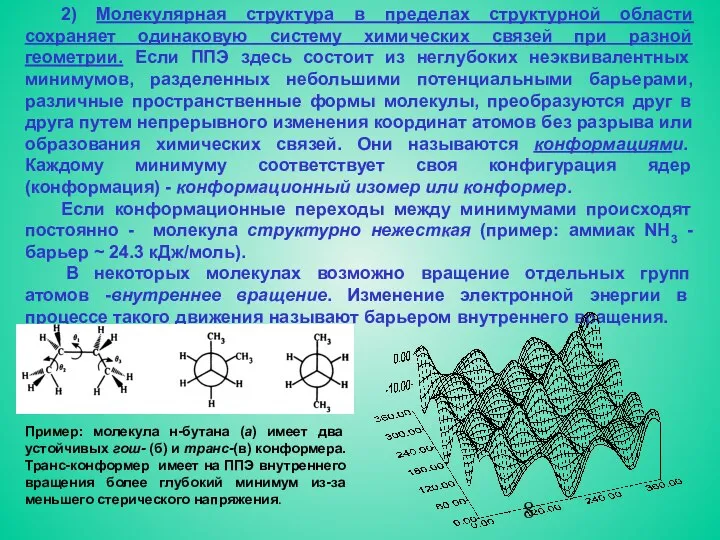
2) Молекулярная структура в пределах структурной области сохраняет одинаковую систему химических
2) Молекулярная структура в пределах структурной области сохраняет одинаковую систему химических

Энергии вращательных барьеров (ккал/моль)
После символа // указан базис, в котором
Энергии вращательных барьеров (ккал/моль)
После символа // указан базис, в котором

2.3 Метод Хартри-Фока для молекул
Зафиксировав ядерную конфигурацию, для анализа электронного поведения
2.3 Метод Хартри-Фока для молекул
Зафиксировав ядерную конфигурацию, для анализа электронного поведения

Полная ХФ энергия молекулы с замкнутыми оболочками
(2.11)
Замечания.
1) В минимизации энергии
Полная ХФ энергия молекулы с замкнутыми оболочками
(2.11)
Замечания.
1) В минимизации энергии

Приближение МО ЛКАО. Уравнения Рутана
Уравнения Хартри-Фока для молекул
Численное решение
МО электрона, который
Приближение МО ЛКАО. Уравнения Рутана
Уравнения Хартри-Фока для молекул
Численное решение
МО электрона, который

(2.15)
(2.16)
(2.17)
(2.18)
Еμ - одно из решений секулярного уравнения
Удобно ввести матрицу зарядов-
(2.16)
(2.17)
(2.18)
Еμ - одно из решений секулярного уравнения
Удобно ввести матрицу зарядов-
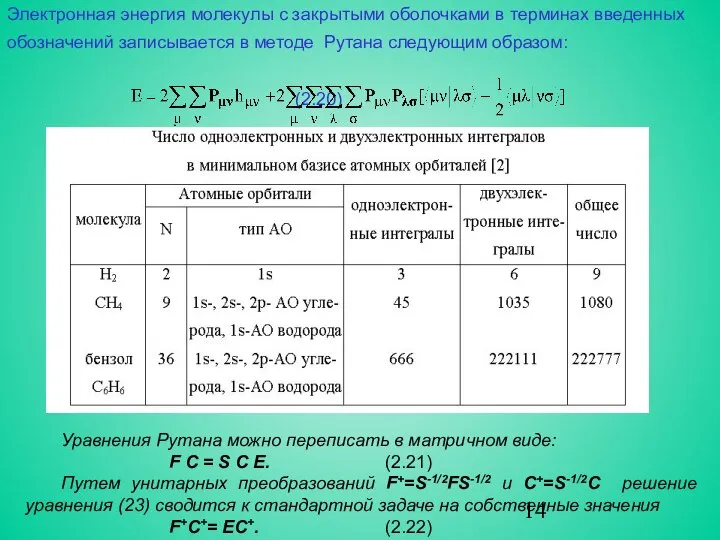
Электронная энергия молекулы с закрытыми оболочками в терминах введенных обозначений записывается
Электронная энергия молекулы с закрытыми оболочками в терминах введенных обозначений записывается
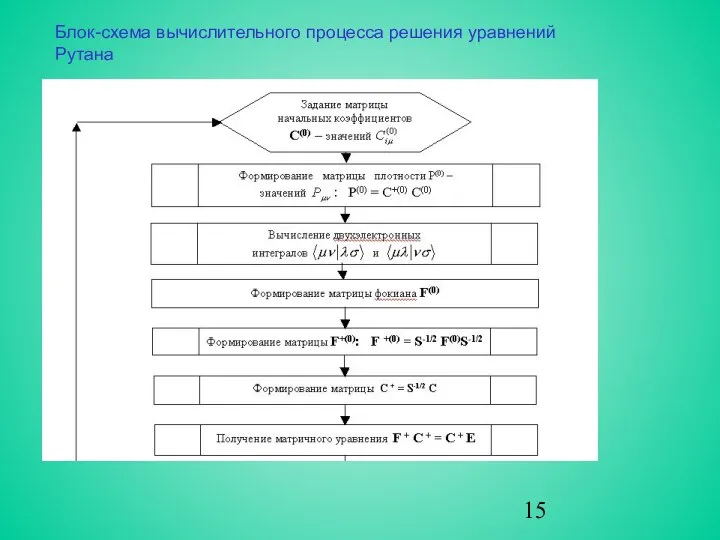
Блок-схема вычислительного процесса решения уравнений Рутана
Блок-схема вычислительного процесса решения уравнений Рутана
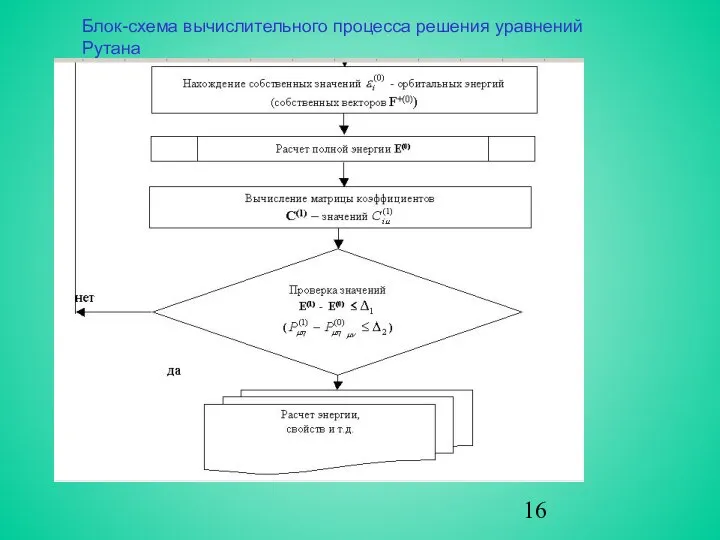
Блок-схема вычислительного процесса решения уравнений Рутана
Блок-схема вычислительного процесса решения уравнений Рутана

Из-за наличия самосогласованного поля уравнения ХФ нелинейны: решения можно получить, лишь
Из-за наличия самосогласованного поля уравнения ХФ нелинейны: решения можно получить, лишь

Многоэлектронная волновая функция и энергии состояний, получаемые с помощью метода Рутана
Многоэлектронная волновая функция и энергии состояний, получаемые с помощью метода Рутана

4. Ограничения метода Хартри-Фока
В ряде молекулярных задач, решаемых методом ХФ, проявляется
4. Ограничения метода Хартри-Фока
В ряде молекулярных задач, решаемых методом ХФ, проявляется
 Спорт во мне
Спорт во мне Демедюк Лілія Мефодіївна- заступник директора з навчально-виховної роботи ЗОШ І-ІІ ст.села Козлів
Демедюк Лілія Мефодіївна- заступник директора з навчально-виховної роботи ЗОШ І-ІІ ст.села Козлів Конст право 11.pptx
Конст право 11.pptx Аналитика: каналы и инструменты
Аналитика: каналы и инструменты ВВС - Тема 2 - к лекции 24.09.2019
ВВС - Тема 2 - к лекции 24.09.2019 Святые и святость
Святые и святость Управление персоналом. Сущность, стратегия, стили управления персоналом. Лекция 4
Управление персоналом. Сущность, стратегия, стили управления персоналом. Лекция 4 О мероприятиях по реализации государственной социальной политики
О мероприятиях по реализации государственной социальной политики The second of February, Thursday
The second of February, Thursday  Тема 3.1: Организация и техника операций в торговле готовой продукцией. Особенности организации и техники по поставке машинотех
Тема 3.1: Организация и техника операций в торговле готовой продукцией. Особенности организации и техники по поставке машинотех Презентация Экологическое страхование
Презентация Экологическое страхование Операторы управления
Операторы управления Кодирование информации
Кодирование информации  Род Сoxiella. Сoxiella burnetii. Q-лихорадка. Название «Q-лихорадка» (от англ. queri - неясный) предложил Э. Деррик, впервые описавший заболевани
Род Сoxiella. Сoxiella burnetii. Q-лихорадка. Название «Q-лихорадка» (от англ. queri - неясный) предложил Э. Деррик, впервые описавший заболевани Жидкоструйные смесительные сопла сита и ситовой анализ
Жидкоструйные смесительные сопла сита и ситовой анализ Стратегия «Казахстан-2050». Новый политический курс состоявшегося государства
Стратегия «Казахстан-2050». Новый политический курс состоявшегося государства Психофізіологічна діагностика у спорті
Психофізіологічна діагностика у спорті МОУ «Крутинская гимназия» Словосочетание в предложении Учитель : Вершинина Т.А
МОУ «Крутинская гимназия» Словосочетание в предложении Учитель : Вершинина Т.А Хочу знать все! - презентация для начальной школы
Хочу знать все! - презентация для начальной школы Урок №31 (112) Тест «Единицы длины»
Урок №31 (112) Тест «Единицы длины»  Рационализация размещения товара на складе на примере предприятия ООО «Гекса – нетканые материалы»
Рационализация размещения товара на складе на примере предприятия ООО «Гекса – нетканые материалы» Определение предмета статистики в широком и узком смысле. Система правовой статистики.
Определение предмета статистики в широком и узком смысле. Система правовой статистики. Аттестационная работа. Программа внеурочной деятельности Пермский край – мой родной край
Аттестационная работа. Программа внеурочной деятельности Пермский край – мой родной край 5S on technical workshop. Бережливое производство
5S on technical workshop. Бережливое производство Антифосфолипидный синдром
Антифосфолипидный синдром Цвет как средство выражения. Теплые и холодные цвета
Цвет как средство выражения. Теплые и холодные цвета Города 1 и 2 уровня
Города 1 и 2 уровня ВКР: Разработка программного модуля проверки АРМ разработчика в среде Navisworks
ВКР: Разработка программного модуля проверки АРМ разработчика в среде Navisworks