- Квантовая химия молекул

Содержание
- 2. молекулярная динамика и метод Монте-Карло неэмпирическая квантовая химия полуэмпирическая квантовая химия квантовая статистическая механика молекулярная механика
- 3. Квантовая химия молекул. Электронная корреляция Метод ХФ дает для энергии диссоциации молекулы Н2 значение 2.65 эВ,
- 4. межъядерное расстояние, Å межъядерное расстояние, Å Зависимость полной энергии молекул F2 и N2 от межъядерного расстояния
- 5. Иллюстрация: Метилен СН2 низколежащее синглетное возбужденное состояние 1A1 Δэкспер=9.2 ккал/моль триплетное основное состояние 3B1 Из-за малости
- 6. Полный метод КВ → точное (в пределах базисного набора) решение электронного уравнения Шредингера. Для больших молекул
- 7. Метод многоконфигурационного взаимодействия (МКВ или МС, англ. MCSCF) - вариационный метод, в котором, в отличие от
- 8. Теория возмущений В ряде задач фигурируют разные по порядку величины: отбрасывая малые величины, задачу можно сильно
- 9. Условие применимости теории возмущений: Н'mn т.е. матричные элементы возмущения должны быть меньше, чем разность энергий невозмущенных
- 10. Если H0 - оператор Фока → теория возмущений Мёллера-Плессета (англ. MPPT), где самая низкая отличная от
- 11. Метод валентных связей Метод валентных связей учитывает, что атомы в молекулах сохраняют во многом свою индивидуальность,
- 12. Электронное строение и свойства системы, таким образом, представляются как среднее по различным ВС, число которых может
- 13. Точность учета электронной корреляции Учет электронной корреляции и увеличение размера и гибкости базисного набора, систематически улучшают
- 14. Расчетные значения длин связей и валентных углов лишь слабо зависят от корреляционных эффектов. Ошибки в длинах
- 15. Расчет энергии диссоциации химических связей Энергия диссоциации химической связи - энергия, необходимая для разрыва молекулы в
- 16. Квантовая химия молекул. Иерархия методов квантовой химии Решение электронного уравнения Шредингера для молекулы: метод Хартри-Фока (+
- 18. Скачать презентацию
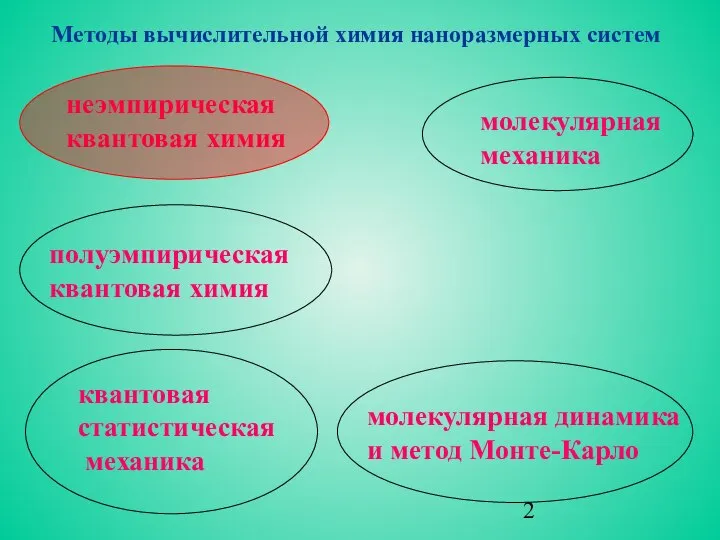
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия

Квантовая химия молекул.
Электронная корреляция
Метод ХФ дает для энергии
Квантовая химия молекул.
Электронная корреляция
Метод ХФ дает для энергии
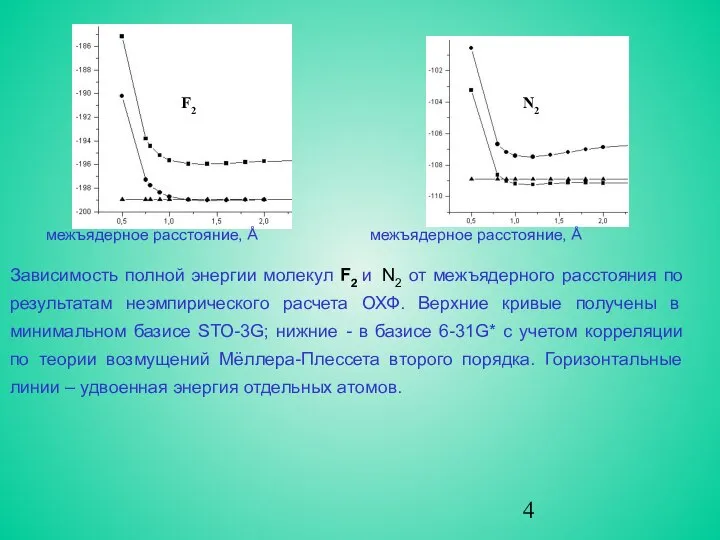
межъядерное расстояние, Å межъядерное расстояние, Å
Зависимость полной энергии молекул F2
межъядерное расстояние, Å межъядерное расстояние, Å
Зависимость полной энергии молекул F2

Иллюстрация: Метилен СН2
низколежащее синглетное
возбужденное состояние 1A1 Δэкспер=9.2 ккал/моль
триплетное основное
Иллюстрация: Метилен СН2
низколежащее синглетное
возбужденное состояние 1A1 Δэкспер=9.2 ккал/моль
триплетное основное

Полный метод КВ → точное (в пределах базисного набора) решение
Полный метод КВ → точное (в пределах базисного набора) решение

Метод многоконфигурационного взаимодействия
(МКВ или МС, англ. MCSCF) - вариационный метод,
Метод многоконфигурационного взаимодействия
(МКВ или МС, англ. MCSCF) - вариационный метод,

Теория возмущений
В ряде задач фигурируют разные по порядку величины: отбрасывая
Теория возмущений
В ряде задач фигурируют разные по порядку величины: отбрасывая

Условие применимости теории возмущений:
Н'mn << ⎢ Еn,0 - Еm,0 ⎢,
Условие применимости теории возмущений:
Н'mn << ⎢ Еn,0 - Еm,0 ⎢,

Если H0 - оператор Фока → теория возмущений Мёллера-Плессета (англ. MPPT),
Если H0 - оператор Фока → теория возмущений Мёллера-Плессета (англ. MPPT),

Метод валентных связей
Метод валентных связей учитывает, что атомы в молекулах сохраняют
Метод валентных связей
Метод валентных связей учитывает, что атомы в молекулах сохраняют

Электронное строение и свойства системы, таким образом, представляются как среднее
Электронное строение и свойства системы, таким образом, представляются как среднее
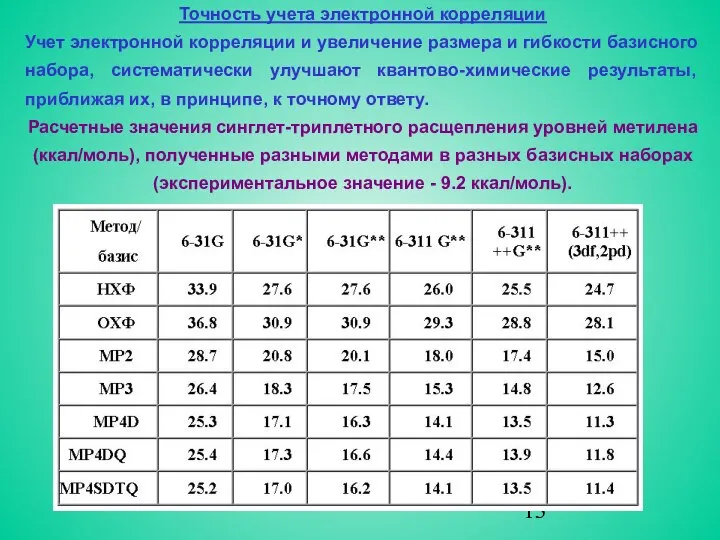
Точность учета электронной корреляции
Учет электронной корреляции и увеличение размера и
Точность учета электронной корреляции
Учет электронной корреляции и увеличение размера и
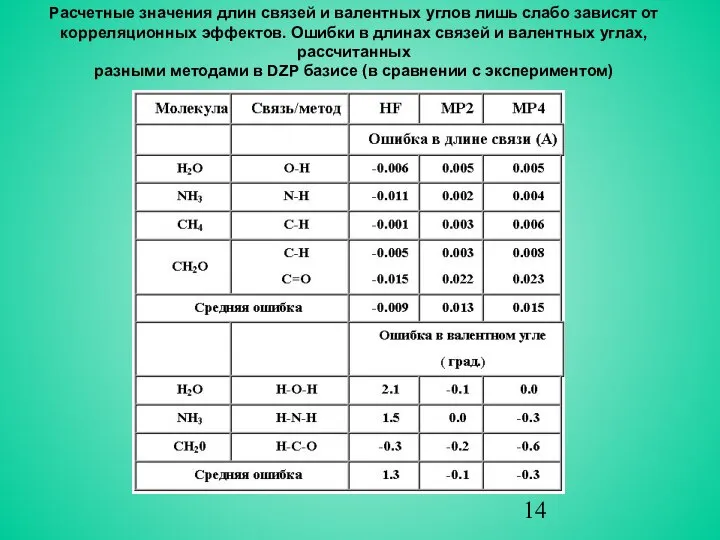
Расчетные значения длин связей и валентных углов лишь слабо зависят от
Расчетные значения длин связей и валентных углов лишь слабо зависят от
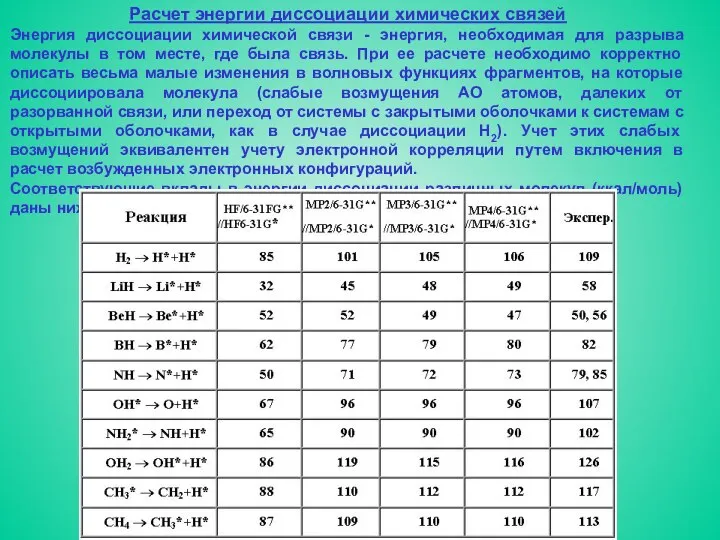
Расчет энергии диссоциации химических связей
Энергия диссоциации химической связи - энергия, необходимая
Расчет энергии диссоциации химических связей
Энергия диссоциации химической связи - энергия, необходимая
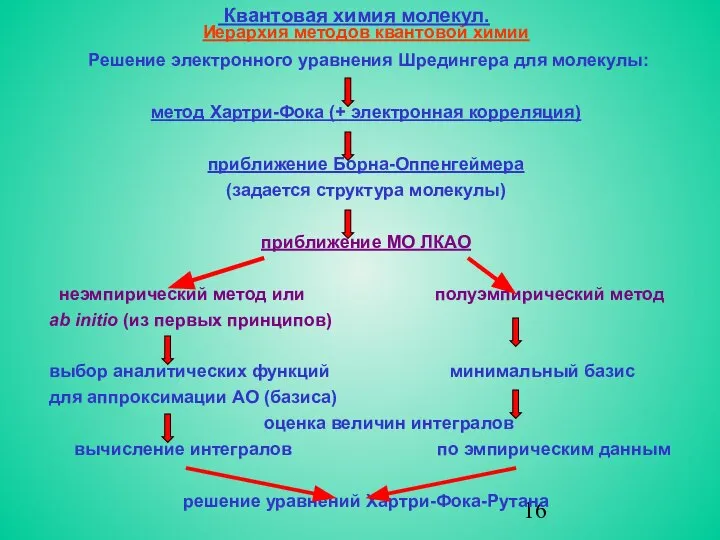
Квантовая химия молекул.
Иерархия методов квантовой химии
Решение электронного уравнения Шредингера
Квантовая химия молекул.
Иерархия методов квантовой химии
Решение электронного уравнения Шредингера
 Verb Tenses Review
Verb Tenses Review Створення креслень в КОМПАС-3D V14/16
Створення креслень в КОМПАС-3D V14/16 Аналого-цифровые преобразователи (АЦП)
Аналого-цифровые преобразователи (АЦП) Психологические и психиатрические подходы к понятию «вменяемости»
Психологические и психиатрические подходы к понятию «вменяемости» Программа түсінігі. Құрылымы. Шамаларды сипаттау
Программа түсінігі. Құрылымы. Шамаларды сипаттау Нормативная база ценообразования в строительстве
Нормативная база ценообразования в строительстве Романтизм или реализм?
Романтизм или реализм? Связь зарядов и потенциалов проводников
Связь зарядов и потенциалов проводников Welsh festivals
Welsh festivals устные вычисления - презентация для начальной школы
устные вычисления - презентация для начальной школы Движение – это жизнь. Валеология
Движение – это жизнь. Валеология ПРОЕКТ «ДЕТСКИЙ САД»
ПРОЕКТ «ДЕТСКИЙ САД» Функционально - планировочная организация города
Функционально - планировочная организация города ЭРА-ГЛОНАСС: общая информация. Государственная система экстренного реагирования при авариях
ЭРА-ГЛОНАСС: общая информация. Государственная система экстренного реагирования при авариях Организация общевойсковых частей и подразделений
Организация общевойсковых частей и подразделений  Тягач «Майло» и его друзья. Тяга. Конструктор
Тягач «Майло» и его друзья. Тяга. Конструктор Как вас зовут? - презентация для начальной школы
Как вас зовут? - презентация для начальной школы Розв’язування крайових задач для звичайних диференціальних рівнянь методом Гальоркіна
Розв’язування крайових задач для звичайних диференціальних рівнянь методом Гальоркіна ПРОЩАНИЕ С АЗБУКОЙ
ПРОЩАНИЕ С АЗБУКОЙ Глава 3. Экономика фирмы 20. Рынок капитала
Глава 3. Экономика фирмы 20. Рынок капитала  Реализм – художественный стиль эпохи
Реализм – художественный стиль эпохи  Молоко Товароведение и экспертиза в таможенном деле
Молоко Товароведение и экспертиза в таможенном деле  Стихи любимого поэта - презентация для начальной школы_
Стихи любимого поэта - презентация для начальной школы_ Основы информационных технологий в профессиональной деятельности агента рекламы
Основы информационных технологий в профессиональной деятельности агента рекламы Электродинамические явления в дуговых печах
Электродинамические явления в дуговых печах ЗАМОК ПРИНЦЕССЫ ОЛЬДЕНБУРГСКОЙ ЗАМЕЧАТЕЛЬНЫЕ МЕСТА ВОРОНЕЖСКОЙ ОБЛАСТИ
ЗАМОК ПРИНЦЕССЫ ОЛЬДЕНБУРГСКОЙ ЗАМЕЧАТЕЛЬНЫЕ МЕСТА ВОРОНЕЖСКОЙ ОБЛАСТИ Краеведческий кружок «Аскольд»
Краеведческий кружок «Аскольд» Материалы к обсуждению закона об образовании
Материалы к обсуждению закона об образовании