- Курс лекций по энзимиологии. Основные вопросы по 3-й лекции. Лекции 4-5

Содержание
- 2. Основные вопросы по 3-й лекции 1. Единицы измерения скорости первого и второго порядка. 2. Размерность констант
- 3. Лекция 4 Лекция 5 1. Необратимые реакции первого и второго порядка. 2. Время полупревращения реакций нулевого
- 4. Определить порядок реакции и значение константы скорости (A0 – начальная концентрация, А-концентрация в момент времени t
- 5. Решение Если построить график в координатах ln A0/A , t, (2,303 lg A0/A = kt) то
- 6. V=[A]k A=B Уравнение скорости первого порядка в интегральной форме 2,303 lg A0/A = kt или lg
- 7. 2A=B График для реакций второго порядка 1/(a-x) = kt +1/a в координатах 1/(a-x), t tg α
- 8. Реакции нулевого порядка V=k Время полупревращения реагента (субстрата) t1/2 для реакций нулевого порядка dx/dt = k
- 9. Зависимость скорости реакции нулевого порядка от концентрации V=k
- 10. Характеристические функции кинетики необратимых химических реакций Порядок 0 (реакции нулевого порядка) Дифференциальное уравнение -d(a-x) / dt
- 11. Характеристические функции кинетики необратимых химических реакций Порядок 1 (реакции первого порядка) V=[A]k A=B Дифференциальное уравнение -d(a-x)
- 12. Характеристические функции кинетики необратимых химических реакций Порядок 2 (реакции второго порядка) 2A=B (A+A) Дифференциальное уравнение -d(a-x)
- 13. Обратимые реакции Химические реакции, которые при одних и тех же условиях могут идти в противоположных направлениях,
- 14. Обратимые реакции mA+nBpC+qD k1 [A]m [B]n = k-1[C]p[D]q Kравн= k1/ k -1 =[C]p[D]q / [A]m [B]n
- 15. Для ферментативных реакций Кравн = k1/ k-1 = [ES] / [E] [S] Kдисссоциации =1/Кравн =Кs= [E]
- 16. Обратимые реакции Константы скорости прямой и обратной реакций k1 и k2 характеризуют 1. химическую природу реагирующих
- 17. Обратимые реакции Kр= k1/ k -1 =[C]p[D]q / [A]m [B]n Kр - константа равновесия, представляющая собой
- 18. Смещение химического равновесия. . Принцип Анри Луи Ле Шателье – Ф. Брауна (1884-1888 гг) (Принцип смещения
- 19. Факторы, влияющие на смещение равновесия Концентрация. При увеличении концентрации одного из реагирующих веществ равновесие смещается в
- 20. Факторы, влияющие на смещение равновенсия Давление (для газовых систем). При увеличении давления равновесие смещается в сторону
- 21. Факторы, влияющие на смещение равновесия Температура. При повышении температуры равновесие смещается в сторону эндотермической реакции (идущей
- 22. *Интегральное определение порядка реакции и констант скоростей обратимых реакций Мономолекулярные реакции первого порядка A B
- 23. * Интегральное определение порядка реакции и констант скоростей обратимых реакций При достижении равновесия v1=v-1 x=x равн
- 24. * Интегральное определение порядка реакции и констант скоростей обратимых реакций dx/dt=(k1a/ xравн)(xравн- x) После интегрирования получим
- 25. * График обратимой реакции первого порядка (xравн /a)ln(xравн/ [xравн- x])=k1t (x равн /a)ln(x равн/ [x равн-
- 26. Константа скорости обратной реакции k-1 может быть вычислена из константы равновесия Кравн по известной k1 (k1
- 27. * Обратимые реакции разных порядков Константы скорости обратимых химических реакций A B Интегральное уравнение (xравн
- 28. *Константы скорости обратимых химических реакций Тип реакции A B+C k1 =[x равн./t (2a-xравн)] ln{[ ax
- 29. * Константы скорости обратимых химических реакций Тип реакции A +B C Если [A]=[B] k1 =[x
- 30. * Константы скорости обратимых химических реакций Тип реакции A +B C Если [A] не равно
- 31. *Реакция n-го порядка Для состояния равновесия различных обратимых реакций в общем случае для n-молекулярной реакции n-го
- 32. Влияние температуры на скорость реакции Скорость любой химической реакции при повышении температуры увеличивается, если при этом
- 33. Изменение константы скорости в зависимости от температуры Влияние температуры на скорость реакции. Правило Вант-Гоффа. При повышении
- 34. Константу скорости реакции при температуре, превышающей исходную на ΔT можно рассчитать k (T+ ΔT) = kT
- 35. Исключением из этого правила является уменьшение скорости ферментативной реакции при температурах, вызывающих денатурацию (необратимые структурные изменения)
- 36. Уравнение Вант-Гоффа выражает скорость изменения величины lnK в зависимости от температуры (уравнение изобары Вант-Гоффа Дифференциальная форма
- 37. Я.Х. Вант-Гофф Якоб Хендрик Вант-Гофф (нидерл. Jacobus Henricus (Henry) van 't Hoff; 30 августа 1852, Роттердам
- 38. Наиболее существенным фактором, определяющим характер влияния температуры на скорость реакции, является кинетическая энергия реагентов, так как
- 39. Казалось бы, такая зависимость связана с увеличением молекулярных столкновений, но при повышении температуры на десять градусов
- 40. Энергия активации Чтобы объяснить наблюдаемые расхождения Сванте Аррениус показал, что влияние температуры сводится к увеличению числа
- 41. Энергия активации Энергия активации - это некоторое избыточное количество энергии (по сравнению со средой), необходимое для
- 42. Kр= k1/ k -1 Поскольку константа равновесия представляет собой отношение констант скоростей прямой и обратной реакции,
- 43. Уравнение Вант-Гоффа выражает скорость изменения величины lnK в зависимости от температуры (уравнение изобары Вант-Гоффа Дифференциальная форма
- 44. Уравнение Аррениуса Согласно Аррениусу, константа скорости химической реакции зависит от температуры экспоненциально k=A e -Eа/(RT) Еа
- 45. Различные формы уравнения Аррениуса ln k =- Ea /RT +ln A ln kT2/kT1 =Ea (T2-T1) /
- 46. Уравнение Аррениуса ln k =- Ea /RT + ln A (А- константа) Эта форма уравнения Аррениуса
- 47. Зависимость логарифма константы скорости от обратной температуры ln k =- Ea /RT +ln A tg α
- 48. Влияние температуры на скорость ферментативной реакции. Определение энергии активации ln k =- Ea /RT +ln A
- 49. Если начальные концентрации всех компонентов реакционной смеси поддерживаются постоянными и изменяют только температуру, то скорости реакций
- 50. Энергия активации в отличие от ΔH (изменение энтальпии ) всегда имеет положительный знак и равна молярному
- 51. Энергия активации Еа представляет собой критическую (минимальную) энергию активации. Выражение e -Eа/(RT) отражает долю молекул, обладающих
- 52. В уравнении k=A e -Eа/(RT) k константа скорости химической реакции А – предэкспоненциальный или вероятностный фактор
- 53. По теории Аррениуса отношение числа активных молекул Nак к общему числу молекул N , т.е. доля
- 54. Профиль реакции. Сравнительные графики зависимости энергии для катализируемой и некатализируемой реакций. Основная причина увеличения скорости реакции
- 55. Процесс активации можно представить как переход через гору из одной долины в другую. Высота горного перевала,
- 56. Задача Во сколько раз увеличится доля активных молекул, если температура возрастет от 00 до 1000 С
- 57. Решение По теории Аррениуса отношение числа активных молекул Nак к общему числу молекул N , т.е.
- 58. k=A e -E/(RT) Энергия активации Ea равна активационной энтальпии Δ H# с обратным знаком Ea =-Δ
- 59. k=A e Δ H # /(RT) Энергия активации Ea всегда имеет положительный знак Активационная энтальпия Δ
- 60. k=A e -Ea/(RT) Если известна зависимость константы скорости реакции от температуры, то энергию активации можно рассчитать
- 61. Теория Эйринга (теория переходных состояний) Кинетический анализ степеней свободы сталкивающихся молекул позволил теоретически вывести уравнение Аррениуса.
- 62. Теория Эйринга (1935 г.) В 1935 г. Эйринг, Эванс и Поляни предложили теорию переходного состояния (теория
- 64. Энергетические кривые для простых ферментативных реакциях при различных относительных величинах констант скоростей
- 65. Изменение свободной энергии при переходе от субстратов и фермента к активированному состоянию ES*, а также при
- 66. Активированным комплексом называют короткоживущее соединение фермента с субстратом ES* (переходное состояние), которое возникает при их сближении
- 67. Связь энтальпии активации с энергией активации ΔH* =Ea –RT ΔH* - энтальпия активации ΔG = -2.3
- 68. Термодинамические уравнения для процесса активации ΔG=RT lnKs или ΔG=-RT lnKравн ΔG*=-RT lnK*равн ΔG*= Δ H*-T ΔS*
- 69. Термодинамические уравнения для процесса активации. Уравнение Эйринга k –константа скорости (произведение двух экспонент) k=kb T/h e
- 70. По теории переходного состояния зависимость ln k от 1/Т дает прямую линию с наклоном –(Δ H*+
- 71. k=kb T/h e Δ S*/R e - ΔH*/RT Уравнение Эйринга: Экспоненциальный член представлен произведением двух экспонент
- 72. Если константу скорости реакции k и величину энергии активации Еа определить экспериментально, то можно рассчитать энтропию
- 74. Скачать презентацию

Основные вопросы по 3-й лекции
1. Единицы измерения скорости первого и второго
Основные вопросы по 3-й лекции
1. Единицы измерения скорости первого и второго

Лекция 4
Лекция 5
1. Необратимые реакции первого и второго порядка.
2. Время
Лекция 4
Лекция 5
1. Необратимые реакции первого и второго порядка.
2. Время

Определить порядок реакции и значение константы скорости (A0 – начальная концентрация,
Определить порядок реакции и значение константы скорости (A0 – начальная концентрация,

Решение
Если построить график в координатах
ln A0/A , t, (2,303
Решение
Если построить график в координатах
ln A0/A , t, (2,303
![V=[A]k A=B Уравнение скорости первого порядка в интегральной форме 2,303 lg](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/686566/slide-5.jpg)
V=[A]k A=B
Уравнение скорости первого порядка в интегральной форме
2,303 lg A0/A =
V=[A]k A=B Уравнение скорости первого порядка в интегральной форме 2,303 lg A0/A =
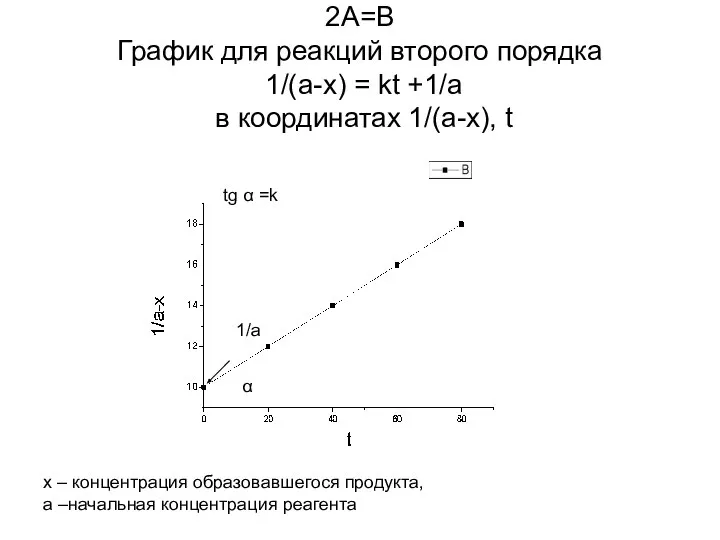
2A=B
График для реакций второго порядка 1/(a-x) = kt +1/a в
2A=B График для реакций второго порядка 1/(a-x) = kt +1/a в

Реакции нулевого порядка V=k
Время полупревращения реагента (субстрата) t1/2 для реакций
Реакции нулевого порядка V=k Время полупревращения реагента (субстрата) t1/2 для реакций
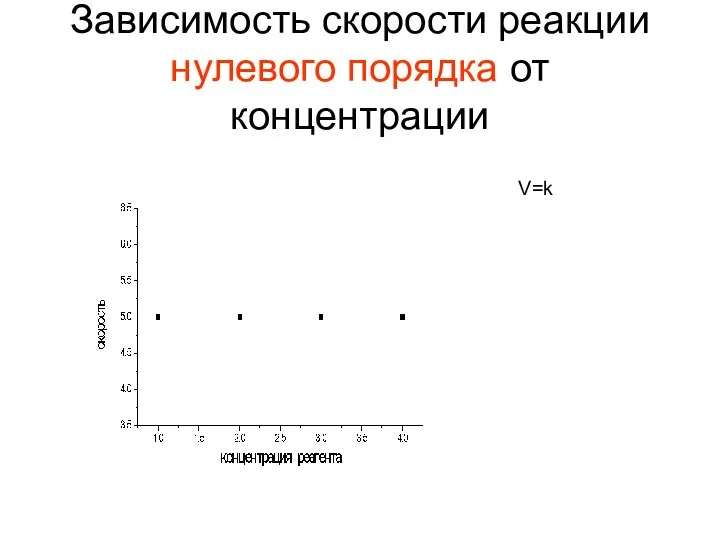
Зависимость скорости реакции нулевого порядка от концентрации
V=k
Зависимость скорости реакции нулевого порядка от концентрации
V=k

Характеристические функции кинетики необратимых химических реакций
Порядок 0 (реакции нулевого порядка)
Дифференциальное уравнение
Характеристические функции кинетики необратимых химических реакций
Порядок 0 (реакции нулевого порядка)
Дифференциальное уравнение

Характеристические функции кинетики необратимых химических реакций
Порядок 1 (реакции первого порядка) V=[A]k A=B
Дифференциальное
Характеристические функции кинетики необратимых химических реакций
Порядок 1 (реакции первого порядка) V=[A]k A=B
Дифференциальное

Характеристические функции кинетики необратимых химических реакций
Порядок 2 (реакции второго порядка) 2A=B
Характеристические функции кинетики необратимых химических реакций
Порядок 2 (реакции второго порядка) 2A=B

Обратимые реакции
Химические реакции, которые при одних и тех же условиях
Обратимые реакции
Химические реакции, которые при одних и тех же условиях
![Обратимые реакции mA+nBpC+qD k1 [A]m [B]n = k-1[C]p[D]q Kравн= k1/ k](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/686566/slide-13.jpg)
Обратимые реакции mA+nBpC+qD
k1 [A]m [B]n = k-1[C]p[D]q
Kравн= k1/ k -1 =[C]p[D]q
Обратимые реакции mA+nBpC+qD
k1 [A]m [B]n = k-1[C]p[D]q
Kравн= k1/ k -1 =[C]p[D]q
![Для ферментативных реакций Кравн = k1/ k-1 = [ES] / [E]](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/686566/slide-14.jpg)
Для ферментативных реакций
Кравн = k1/ k-1 = [ES] / [E]
Для ферментативных реакций
Кравн = k1/ k-1 = [ES] / [E]

Обратимые реакции
Константы скорости прямой и обратной реакций k1 и k2 характеризуют
Обратимые реакции
Константы скорости прямой и обратной реакций k1 и k2 характеризуют
![Обратимые реакции Kр= k1/ k -1 =[C]p[D]q / [A]m [B]n Kр](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/686566/slide-16.jpg)
Обратимые реакции
Kр= k1/ k -1 =[C]p[D]q / [A]m [B]n
Kр - константа
Обратимые реакции
Kр= k1/ k -1 =[C]p[D]q / [A]m [B]n
Kр - константа

Смещение химического равновесия.
.
Принцип Анри Луи Ле Шателье – Ф. Брауна
Смещение химического равновесия.
.
Принцип Анри Луи Ле Шателье – Ф. Брауна

Факторы, влияющие на смещение равновесия
Концентрация. При увеличении концентрации одного из реагирующих
Факторы, влияющие на смещение равновесия
Концентрация. При увеличении концентрации одного из реагирующих

Факторы, влияющие на смещение равновенсия
Давление (для газовых систем). При увеличении давления
Факторы, влияющие на смещение равновенсия
Давление (для газовых систем). При увеличении давления

Факторы, влияющие на смещение равновесия
Температура. При повышении температуры равновесие смещается в
Факторы, влияющие на смещение равновесия
Температура. При повышении температуры равновесие смещается в

*Интегральное определение порядка реакции и констант скоростей обратимых реакций
Мономолекулярные реакции первого
*Интегральное определение порядка реакции и констант скоростей обратимых реакций
Мономолекулярные реакции первого

* Интегральное определение порядка реакции и констант скоростей обратимых реакций
При достижении
* Интегральное определение порядка реакции и констант скоростей обратимых реакций
При достижении

* Интегральное определение порядка реакции и констант скоростей обратимых реакций
dx/dt=(k1a/
* Интегральное определение порядка реакции и констант скоростей обратимых реакций
dx/dt=(k1a/
![* График обратимой реакции первого порядка (xравн /a)ln(xравн/ [xравн- x])=k1t (x](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/686566/slide-24.jpg)
* График обратимой реакции первого порядка
(xравн /a)ln(xравн/ [xравн- x])=k1t
(x равн /a)ln(x
* График обратимой реакции первого порядка
(xравн /a)ln(xравн/ [xравн- x])=k1t
(x равн /a)ln(x

Константа скорости обратной реакции
k-1 может быть вычислена из константы равновесия
Константа скорости обратной реакции
k-1 может быть вычислена из константы равновесия

* Обратимые реакции разных порядков
Константы скорости обратимых химических реакций
A B
Интегральное
* Обратимые реакции разных порядков
Константы скорости обратимых химических реакций
A B
Интегральное

*Константы скорости обратимых химических реакций
Тип реакции
A B+C
k1 =[x
*Константы скорости обратимых химических реакций
Тип реакции
A B+C
k1 =[x

* Константы скорости обратимых химических реакций
Тип реакции
A +B C
Если
* Константы скорости обратимых химических реакций
Тип реакции
A +B C
Если

* Константы скорости обратимых химических реакций
Тип реакции
A +B C
Если
* Константы скорости обратимых химических реакций
Тип реакции
A +B C
Если

*Реакция n-го порядка
Для состояния равновесия различных обратимых реакций в общем случае
*Реакция n-го порядка
Для состояния равновесия различных обратимых реакций в общем случае

Влияние температуры на скорость реакции
Скорость любой химической реакции при повышении температуры
Влияние температуры на скорость реакции
Скорость любой химической реакции при повышении температуры

Изменение константы скорости в зависимости от температуры
Влияние температуры на скорость реакции.
Изменение константы скорости в зависимости от температуры
Влияние температуры на скорость реакции.

Константу скорости реакции при температуре, превышающей исходную на ΔT можно рассчитать
Константу скорости реакции при температуре, превышающей исходную на ΔT можно рассчитать

Исключением из этого правила является уменьшение скорости ферментативной реакции при температурах,
Исключением из этого правила является уменьшение скорости ферментативной реакции при температурах,

Уравнение Вант-Гоффа
выражает скорость изменения величины lnK в зависимости от температуры (уравнение
Уравнение Вант-Гоффа выражает скорость изменения величины lnK в зависимости от температуры (уравнение

Я.Х. Вант-Гофф
Якоб Хендрик Вант-Гофф (нидерл. Jacobus Henricus (Henry) van 't Hoff; 30
Я.Х. Вант-Гофф
Якоб Хендрик Вант-Гофф (нидерл. Jacobus Henricus (Henry) van 't Hoff; 30

Наиболее существенным фактором, определяющим характер влияния температуры на скорость реакции, является
Наиболее существенным фактором, определяющим характер влияния температуры на скорость реакции, является

Казалось бы, такая зависимость связана с увеличением молекулярных столкновений, но при
Казалось бы, такая зависимость связана с увеличением молекулярных столкновений, но при

Энергия активации
Чтобы объяснить наблюдаемые расхождения Сванте Аррениус показал, что влияние температуры
Энергия активации
Чтобы объяснить наблюдаемые расхождения Сванте Аррениус показал, что влияние температуры

Энергия активации
Энергия активации - это некоторое избыточное количество энергии (по сравнению
Энергия активации
Энергия активации - это некоторое избыточное количество энергии (по сравнению

Kр= k1/ k -1
Поскольку константа равновесия представляет собой отношение констант скоростей
Kр= k1/ k -1
Поскольку константа равновесия представляет собой отношение констант скоростей

Уравнение Вант-Гоффа
выражает скорость изменения величины lnK в зависимости от температуры (уравнение
Уравнение Вант-Гоффа выражает скорость изменения величины lnK в зависимости от температуры (уравнение

Уравнение Аррениуса
Согласно Аррениусу, константа скорости химической реакции зависит от температуры экспоненциально
k=A
Уравнение Аррениуса
Согласно Аррениусу, константа скорости химической реакции зависит от температуры экспоненциально
k=A

Различные формы уравнения Аррениуса
ln k =- Ea /RT +ln A
ln
Различные формы уравнения Аррениуса
ln k =- Ea /RT +ln A
ln

Уравнение Аррениуса
ln k =- Ea /RT + ln A (А- константа)
Эта
Уравнение Аррениуса
ln k =- Ea /RT + ln A (А- константа)
Эта
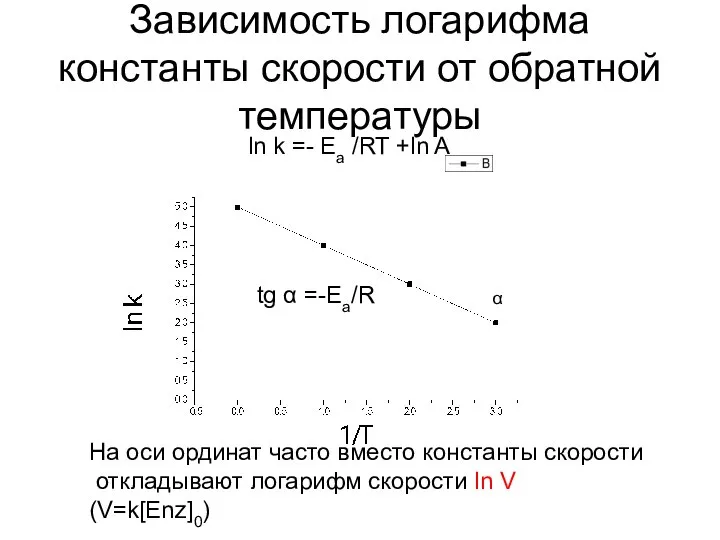
Зависимость логарифма константы скорости от обратной температуры
ln k =- Ea /RT
Зависимость логарифма константы скорости от обратной температуры
ln k =- Ea /RT
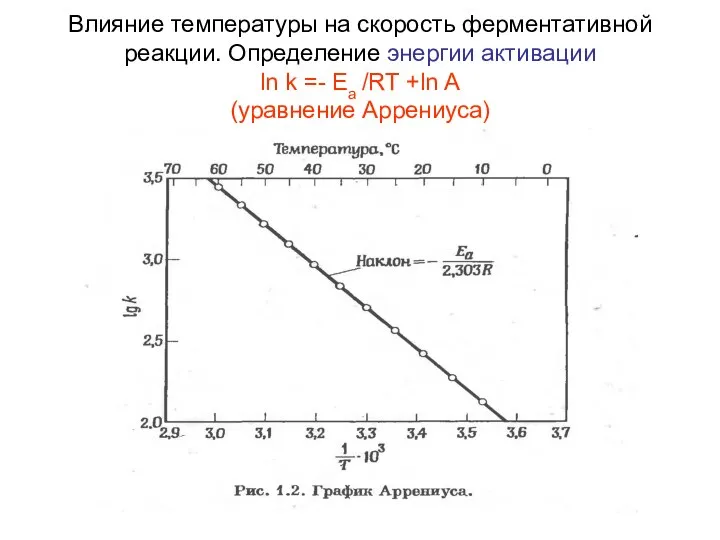
Влияние температуры на скорость ферментативной реакции. Определение энергии активации
ln k =-
Влияние температуры на скорость ферментативной реакции. Определение энергии активации ln k =-

Если начальные концентрации всех компонентов реакционной смеси поддерживаются постоянными и изменяют
Если начальные концентрации всех компонентов реакционной смеси поддерживаются постоянными и изменяют

Энергия активации в отличие от ΔH (изменение энтальпии ) всегда имеет
Энергия активации в отличие от ΔH (изменение энтальпии ) всегда имеет

Энергия активации Еа представляет собой критическую (минимальную) энергию активации.
Выражение e
Энергия активации Еа представляет собой критическую (минимальную) энергию активации.
Выражение e

В уравнении k=A e -Eа/(RT)
k константа скорости химической реакции
А –
В уравнении k=A e -Eа/(RT)
k константа скорости химической реакции
А –

По теории Аррениуса отношение числа активных молекул Nак к общему числу
По теории Аррениуса отношение числа активных молекул Nак к общему числу
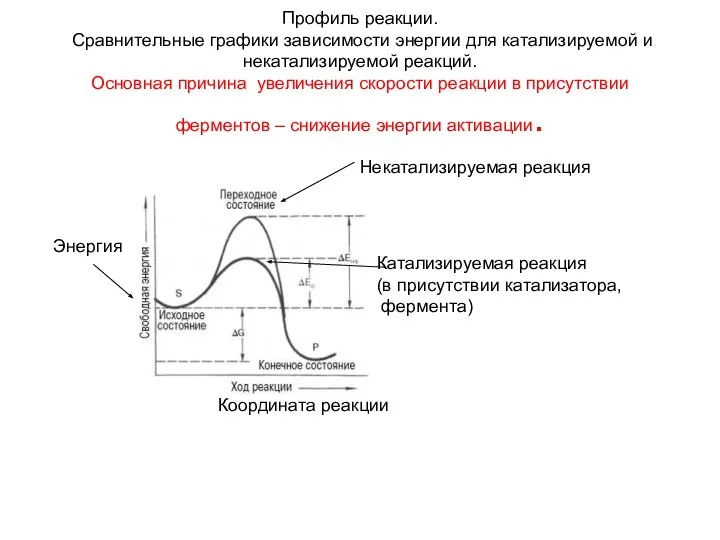
Профиль реакции.
Сравнительные графики зависимости энергии для катализируемой и некатализируемой реакций.
Основная
Профиль реакции. Сравнительные графики зависимости энергии для катализируемой и некатализируемой реакций. Основная

Процесс активации можно представить как переход через гору из одной долины
Процесс активации можно представить как переход через гору из одной долины

Задача
Во сколько раз увеличится доля активных молекул, если температура возрастет от
Задача
Во сколько раз увеличится доля активных молекул, если температура возрастет от

Решение
По теории Аррениуса отношение числа активных молекул Nак к общему числу
Решение
По теории Аррениуса отношение числа активных молекул Nак к общему числу

k=A e -E/(RT)
Энергия активации Ea равна активационной энтальпии Δ H# с
k=A e -E/(RT)
Энергия активации Ea равна активационной энтальпии Δ H# с

k=A e Δ H # /(RT)
Энергия активации Ea всегда имеет положительный
k=A e Δ H # /(RT)
Энергия активации Ea всегда имеет положительный

k=A e -Ea/(RT)
Если известна зависимость константы скорости реакции от температуры, то
k=A e -Ea/(RT)
Если известна зависимость константы скорости реакции от температуры, то

Теория Эйринга (теория переходных состояний)
Кинетический анализ степеней свободы сталкивающихся молекул позволил
Теория Эйринга (теория переходных состояний)
Кинетический анализ степеней свободы сталкивающихся молекул позволил

Теория Эйринга (1935 г.)
В 1935 г. Эйринг, Эванс и Поляни предложили
Теория Эйринга (1935 г.) В 1935 г. Эйринг, Эванс и Поляни предложили
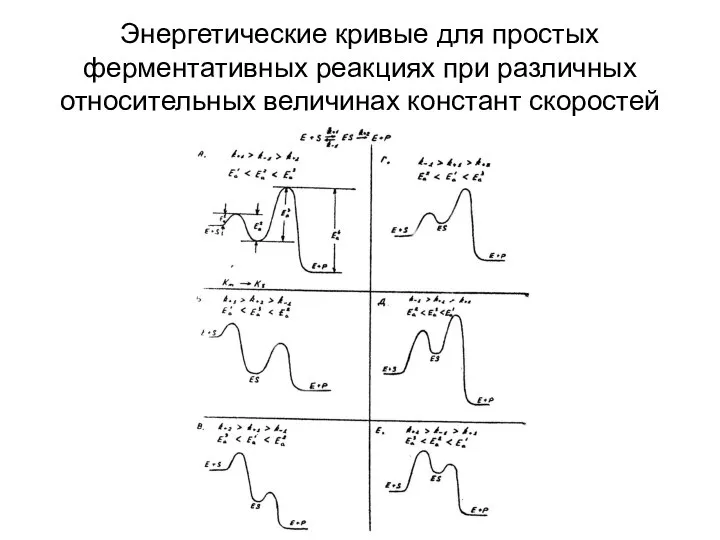
Энергетические кривые для простых ферментативных реакциях при различных относительных величинах констант
Энергетические кривые для простых ферментативных реакциях при различных относительных величинах констант

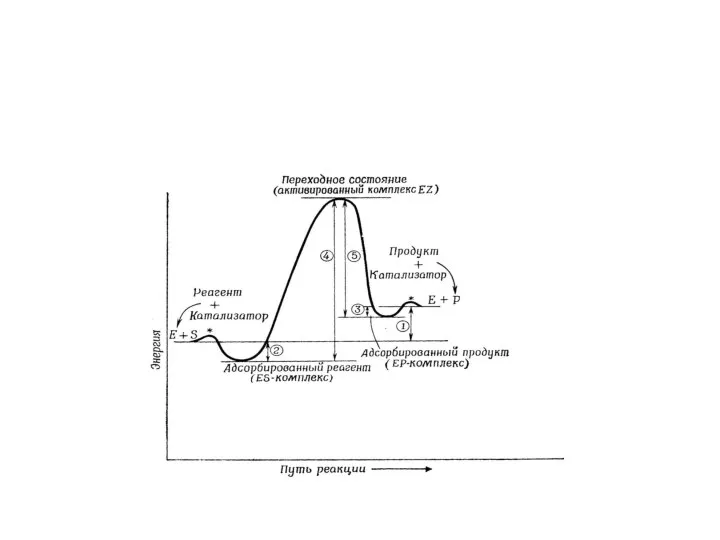
Изменение свободной энергии при переходе от субстратов и фермента к активированному
Изменение свободной энергии при переходе от субстратов и фермента к активированному

Активированным комплексом называют короткоживущее соединение фермента с субстратом ES* (переходное состояние),
Активированным комплексом называют короткоживущее соединение фермента с субстратом ES* (переходное состояние),

Связь энтальпии активации с энергией активации
ΔH* =Ea –RT
ΔH* - энтальпия активации
ΔG
Связь энтальпии активации с энергией активации
ΔH* =Ea –RT
ΔH* - энтальпия активации
ΔG

Термодинамические уравнения для процесса активации
ΔG=RT lnKs или ΔG=-RT lnKравн
ΔG*=-RT lnK*равн
Термодинамические уравнения для процесса активации
ΔG=RT lnKs или ΔG=-RT lnKравн
ΔG*=-RT lnK*равн

Термодинамические уравнения для процесса активации. Уравнение Эйринга
k –константа скорости (произведение
Термодинамические уравнения для процесса активации. Уравнение Эйринга
k –константа скорости (произведение

По теории переходного состояния зависимость ln k от 1/Т дает прямую
По теории переходного состояния зависимость ln k от 1/Т дает прямую

k=kb T/h e Δ S*/R e - ΔH*/RT
Уравнение Эйринга:
Экспоненциальный член представлен
k=kb T/h e Δ S*/R e - ΔH*/RT
Уравнение Эйринга:
Экспоненциальный член представлен

Если константу скорости реакции k и величину энергии активации Еа определить
Если константу скорости реакции k и величину энергии активации Еа определить
 Тема урока: Гигиена питания и предупреждение желудочно-кишечных заболеваний.
Тема урока: Гигиена питания и предупреждение желудочно-кишечных заболеваний. Презентация на тему "Гистологическое строение твердых тканей зуба" - скачать презентации по Биологии
Презентация на тему "Гистологическое строение твердых тканей зуба" - скачать презентации по Биологии Цветок. Орган семейного размножения растений
Цветок. Орган семейного размножения растений Книжная панорама: Неизвестный Мичурин
Книжная панорама: Неизвестный Мичурин Агранулоцити. Моноцити і лімфоцити
Агранулоцити. Моноцити і лімфоцити Тема: Полимерные трохофорные: Тип Annelida (Кольчатые черви) Тип Pogonophora (Погонофоры)
Тема: Полимерные трохофорные: Тип Annelida (Кольчатые черви) Тип Pogonophora (Погонофоры)  Хрящевые рыбы
Хрящевые рыбы Использование техник виртуальной реальности в обучении школьной биологии
Использование техник виртуальной реальности в обучении школьной биологии Методы исследования в биологии. Лабораторное оборудование
Методы исследования в биологии. Лабораторное оборудование 3D-био принтирование. НИО (медико-биологических исследований)
3D-био принтирование. НИО (медико-биологических исследований) Паразитические растения и грибы
Паразитические растения и грибы Презентация Видообразование
Презентация Видообразование Учебно-исследовательская работа на тему Клюква и витамин С
Учебно-исследовательская работа на тему Клюква и витамин С Информационный опорный конспект Липиды
Информационный опорный конспект Липиды Познательные процессы
Познательные процессы Антибиотические отношения. Конкуренция
Антибиотические отношения. Конкуренция Презентация по биологии Отряд Стрекозы
Презентация по биологии Отряд Стрекозы  Происхождение человека
Происхождение человека Консультация к ЕГЭ. Аутосомно-доминантный тип наследования
Консультация к ЕГЭ. Аутосомно-доминантный тип наследования Муниципальное Автономное Образовательное Учреждение Лицей №62 Тема: БИОЛОГИЯ – наука о живой природе
Муниципальное Автономное Образовательное Учреждение Лицей №62 Тема: БИОЛОГИЯ – наука о живой природе  Грудная клетка. Мышцы туловища
Грудная клетка. Мышцы туловища Презентация Ученика 7«А»класса Пилюченок Фёдора.
Презентация Ученика 7«А»класса Пилюченок Фёдора. Технические вредители. (Лекция 14)
Технические вредители. (Лекция 14) Окружающий мир 3 класс Тема: Кровь и кровообращение
Окружающий мир 3 класс Тема: Кровь и кровообращение Покровные органы. Терморегуляция. Выделение
Покровные органы. Терморегуляция. Выделение Физиология и основные характеристики возбудимых тканей
Физиология и основные характеристики возбудимых тканей Пространственная организация генома. Механизм транскрипции, синтез РНК по матрице ДНК. (Лекции 5-6)
Пространственная организация генома. Механизм транскрипции, синтез РНК по матрице ДНК. (Лекции 5-6) Влияние распорядка учебного дня на работоспособность и концентрацию и оптимизация временных затрат на учебный процесс
Влияние распорядка учебного дня на работоспособность и концентрацию и оптимизация временных затрат на учебный процесс