- Второе начало термодинамики

Содержание
- 2. так как , то т.е. ΔSц.к.= 0 или т.е. S – константа. Это выражение называют равенство
- 3. Таким образом ΔSнеобр > 0 или Это называют – неравенство Клаузиуса. При любом необратимом процессе в
- 4. 5. Второе начало термодинамики Термодинамика, как мы уже поняли, это наука о тепловых процессах, о превращении
- 5. Рассмотрим схему теплового двигателя. От термостата с более высокой температурой Т1, называемого нагревателем за цикл отнимается
- 6. Невозможно создание вечного двигателя Второго рода подтверждается вторым началом термодинамики: 1. Невозможен процесс, единственным результатом которого
- 7. Энтропия замкнутой системы при любых, происходивших в ней процессах, не может убывать (или увеличиваться или оставаться
- 8. 6. Свободная и связанная энергии Как мы только что записали, в обратимом процессе: δA= -(dU -
- 9. Т.е. Аизот=F1–F2, следовательно, свободная энергия есть та работа, которая могло бы совершить тело в обратимом изотермическом
- 10. В термодинамике есть еще понятие – энергетическая потеря в изолированной системе где Tmin − температура окружающей
- 11. 7. Статистический смысл энтропии Посмотрим на энтропию с другой стороны. Макросостояние – это состояние вещества характе-ризующее
- 12. Состояние макроскопического тела (т. е. тела, образованного огромным количеством молекул) может быть задано с помощью объема,
- 13. Начнем со случая, когда полное число молекул равно четырем (рис. 1). Каждая молекула с равной вероятностью
- 14. Таблица 1
- 15. Из рассмотренного примера вытекает, что все микросостояния данной системы равновероятны, вследствие чего статистический вес оказывается пропорциональным
- 16. Домножив и разделив это число на (N-n)!, получим выражение Однако не все z способов приводят к
- 17. (27.2) Легко убедиться в том, что Ω(2,4-2) = 6, а Ω(1,4-1) = 4 (см. табл. 1).
- 18. Случайные отклонения значений какой-либо физической величины х от ее среднего значения называются ф л у к
- 19. Вычислим на основании данных табл. 1 относительную флуктуацию числа молекул в левой половине сосуда. В табл.
- 20. Составив пропорцию, аналогичную (27.7), для N-4 и N=1020, получим относительную флуктуацию (о. ф.) числа молекул в
- 21. Система, находящаяся в равновесном состоянии, время от времени самопроизвольно отклоняется от равновесия. Однако эти отклонения являются
- 28. Термодинамической вероятностью или статисти-ческим весом макросостояния W − называется число макросостояний, которым она может быть осуществлена
- 29. Вероятность сложного события, есть W =W1∙W2, где W1 – первое состояние; W2 – второе состояние. Поэтому
- 30. Например, в ящике черные и белые шары. Они порознь, есть порядок и W невелика. После встряхивания
- 31. На этих рассуждениях Клаузиус в 1877 году и выдвинул гипотезу о тепловой смерти Вселенной (о ней
- 32. 8. Третье начало термодинамики Первое и Второе начало термодинамики не позволяет определить значение энтропии при абсолютном
- 33. Обычно его формулируют следующим образом: энтропия любой равновесной системы при абсолютном нуле температуры может быть равна
- 34. При T = 0, внутренняя энергия и тепловая функция системы прекращают зависеть от температуры, кроме того
- 35. Третье начало термодинамики иногда формулируют следующим способом: при абсолютном нуле температуры любые изменения термодинамической системы происходят
- 36. Следствием Третьего начала является, то что невозможно охладить тело до абсолютного нуля (принцип недостижимости абсолютного нуля
- 37. Лекция 29. Реальные газы Реальные газы Уравнение Ван-дер-Ваальса Изотермы уравнения Ван-дер- Ваальса Внутренняя энергия газа Ван-дер-Ваальса
- 38. 7.1. Реальные газы Как известно, уравнение состояния устанавливает функциональную связь между давлением Р, объемом V, температурой
- 39. Самым простым и известным уравнением состояния является уравнение состояния идеального газа: , где R – универсальная
- 40. Для газов с низкой температурой сжиже-ния (He, H2, Ne и даже N2, O2, Ar, CO, CH4)
- 41. Наибольшее распространение вследствие простоты и физической наглядности получило уравнение Ван-дер-Ваальса (1873). Первая поправка в уравнении состояния
- 42. При понижении температуры межмолекулярное взаимодействие в реальных газах приводит к конденсации (образование жидкости). Межмолекулярное притяжение эквивалентно
- 43. Ван-дер-Ваальс в 1873 г. Дал функциональную интерпретацию внутреннего давления. Согласно модели Ван-дер-Ваальса, силы притяжения между молекулами
- 44. С учетом этих соображений уравнение состояния идеального газа преобразуется в уравнение Ван-дер-Ваальса: , или для одного
- 45. Помимо Нобелевской премии, Ван-дер-Ваальс получил почетную докторскую степень Кембриджского университета. Кроме того, он являлся членом Нидерландской
- 46. Реальные газы – газы, свойства которых зависят от взаимодействия молекул. В обычных условиях, когда средняя потенциальная
- 47. Я.Д. Ван-дер-Ваальс для объяснения свойств реальных газов и жидкостей, предположил, что на малых расстояниях между молекулами
- 48. Ориентационные силы действуют между полярными молекулами – молекулами, обладающими дипольными или квадрупольными моментами. Сила притяжения между
- 49. Рисунок 7.1
- 50. Среднее значение потенциальной энергии ориентационного межмолекулярного взаимодействия равно Uор(r) ~ Р1 Р2 r−6, где p1, p2
- 51. Индукционные (поляризационные) силы действуют между полярной и неполярной молекулами, а также между полярными молекулами. Полярная молекула
- 52. Индукционные силы убывают по тому же закону, что и ориентационные F инд ~ r –7. Дисперсионное
- 53. Данное взаимодействие называется дисперсионным, его энергия определяется поляризуемостью молекул α1, α2: U(r) ~ α1α2 r –6,
- 54. Отметим, что все три силы и энергии одинаковым образом убывают с расстоянием F = Fор +
- 55. Силы отталкивания действуют между молекулами на очень малых расстояниях, когда происходит взаимодействие электронных оболочек атомов, входящих
- 56. К хорошему согласию с данными экспериментов приводит допущение, что потенциальная энергия сил отталкивания возрастает с уменьшением
- 57. U( r) = – ar –6 + br –12 Рисунок 7.2
- 58. Глубина потенциала равна U(rmin) = –a2/4b при rmin = (2b/a)1/6 – расстоянии, соответствующем наибольшей энергии связи
- 59. 7.2. Уравнение Ван-дер-Ваальса Уравнение Ван-дер-Ваальса – одно из первых уравнений состояния реального газа, которое было предложено
- 60. Учтем влияние конечных размеров молекул на уравнение состояния реального газа. Давление определяется средней кинетической энергией теплового
- 61. В результате в сосуде, содержащем N молекул конечных размеров, область объемом (N/2)4π(2r)3/3 = 4NVмолек (Vмолек =
- 62. Объем, доступный точечным молекулам, будет равен V − b, а давление, оказываемое на стенки сосуда, определяется
- 63. Для ν = m/μ молей газа уравнение состояния газа с учетом конечного размера молекул примет вид
- 64. Рассмотрим теперь влияние сил притяжения на уравнение состояния идеального газа. Будем считать для простоты частицы газа
- 65. Это обусловлено тем, что в то время как в объеме газа действие сил притяжения между молекулами
- 66. Рисунок 7.3
- 67. Дополнительное внутреннее давление пропорционально числу частиц, приходящихся на единицу площади границы nS и силе взаимодействия этих
- 68. В результате избыточное внутреннее давление Pi (i − intrinsic) будет пропорционально квадрату концентрации числа частиц Pi
- 69. С учетом внутреннего давления уравнение состояния примет вид P + Pi = nkT. Причем давление Pi
- 70. Полученное уравнение с учетом выражения для Pi переходит в новое уравнение состояния реального газа при наличии
- 71. Данное уравнение справедливо при условии νb
- 72. Константы Ван-дер-Ваальса и критические данные Таблица 7.1.
- 73. Примечание. Константы а и b выбраны таким образом, чтобы получить оптимальное согласование уравнения Ван-дер-Ваальса с измеренными
- 74. 7.3. Изотермы уравнения Ван-дер-Ваальса Проанализируем изотермы уравнения Ван-дер-Ваальса – зависимости Р от V для реального газа
- 75. Поскольку данное уравнение имеет третью степень относительно V, а коэффициенты при V действительны, то оно имеет
- 76. Рисунок 7.4
- 77. Изотерма при Ткр, которая разделяет немонотонные T Tкр изотермы, соответствует изотерме при критической температуре. При температуре
- 78. При температуре газа ниже критической такая однозначность исчезает, а это означает возможность перехода вещества из газообразного
- 79. При квазистатическом сжатии, начиная с точки G, система распадается на 2 фазы – жидкость и газ,
- 80. Критическую точку K мы определили как точку перегиба критической изотермы, в которой касательная к изотерме горизонтальна
- 81. 7.4. Внутренняя энергия газа Ван-дер-Ваальса Энергия одного моля газа Ван-дер-Ваальса слагается из внутренней энергии молекул, составляющих
- 82. Потенциальная энергия притяжения молекул равна работе, необходимой для разведения молекул на бесконечное расстояние друг от друга.
- 83. Знак «минус» указывает на то, что между молекулами действуют силы притяжения; Vm – молярный объем, Vm
- 84. Принципиальное значение уравнения Ван-дер-Ваальса определяется следующими обстоятельствами: 1) уравнение было получено из модельных представлений о свойствах
- 85. Причиной недостаточной точности уравнения Ван-дер-Ваальс считал ассоциацию молекул в газовой фазе, которую не удается описать, учитывая
- 87. Скачать презентацию

так как , то
т.е. ΔSц.к.= 0 или
т.е. S – константа. Это
так как , то
т.е. ΔSц.к.= 0 или
т.е. S – константа. Это

Таким образом ΔSнеобр > 0 или
Это называют – неравенство Клаузиуса.
При любом
Таким образом ΔSнеобр > 0 или
Это называют – неравенство Клаузиуса.
При любом

5. Второе начало термодинамики
Термодинамика, как мы уже поняли, это наука о
5. Второе начало термодинамики
Термодинамика, как мы уже поняли, это наука о

Рассмотрим схему теплового двигателя. От термостата с более высокой температурой Т1,
Рассмотрим схему теплового двигателя. От термостата с более высокой температурой Т1,

Невозможно создание вечного двигателя Второго рода подтверждается вторым началом термодинамики:
1. Невозможен
Невозможно создание вечного двигателя Второго рода подтверждается вторым началом термодинамики:
1. Невозможен

Энтропия замкнутой системы при любых, происходивших в ней процессах, не может
Энтропия замкнутой системы при любых, происходивших в ней процессах, не может

6. Свободная и связанная энергии
Как мы только что записали, в обратимом
6. Свободная и связанная энергии
Как мы только что записали, в обратимом

Т.е. Аизот=F1–F2, следовательно, свободная энергия есть та работа, которая могло бы
Т.е. Аизот=F1–F2, следовательно, свободная энергия есть та работа, которая могло бы

В термодинамике есть еще понятие – энергетическая потеря в изолированной системе
где
В термодинамике есть еще понятие – энергетическая потеря в изолированной системе
где

7. Статистический смысл энтропии
Посмотрим на энтропию с другой стороны. Макросостояние –
7. Статистический смысл энтропии
Посмотрим на энтропию с другой стороны. Макросостояние –

Состояние макроскопического тела (т. е. тела, образованного огромным количеством молекул) может
Состояние макроскопического тела (т. е. тела, образованного огромным количеством молекул) может

Начнем со случая, когда полное число молекул равно четырем (рис. 1).
Начнем со случая, когда полное число молекул равно четырем (рис. 1).
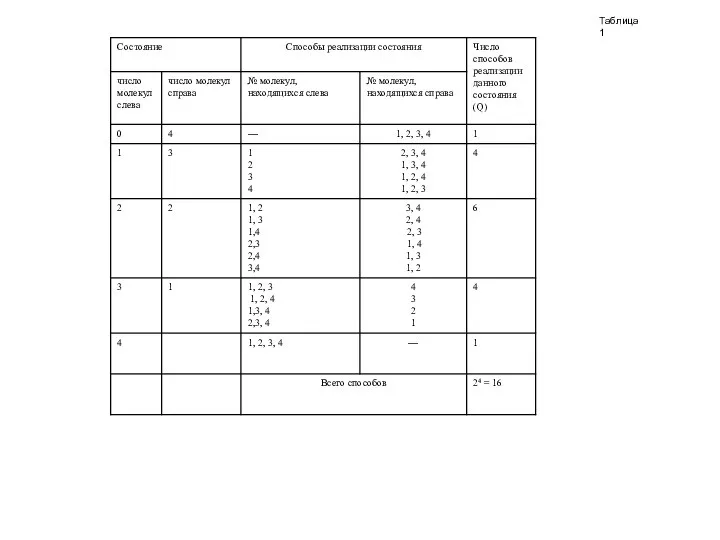
Таблица 1
Таблица 1

Из рассмотренного примера вытекает, что все микросостояния данной системы равновероятны, вследствие
Из рассмотренного примера вытекает, что все микросостояния данной системы равновероятны, вследствие

Домножив и разделив это число на (N-n)!, получим выражение
Однако не все
Домножив и разделив это число на (N-n)!, получим выражение
Однако не все

(27.2)
Легко убедиться в том, что Ω(2,4-2) = 6, а Ω(1,4-1) =
(27.2)
Легко убедиться в том, что Ω(2,4-2) = 6, а Ω(1,4-1) =

Случайные отклонения значений какой-либо физической величины х от ее среднего значения
Случайные отклонения значений какой-либо физической величины х от ее среднего значения

Вычислим на основании данных табл. 1 относительную флуктуацию числа молекул в
Вычислим на основании данных табл. 1 относительную флуктуацию числа молекул в

Составив пропорцию, аналогичную (27.7), для N-4 и N=1020, получим относительную флуктуацию
Составив пропорцию, аналогичную (27.7), для N-4 и N=1020, получим относительную флуктуацию

Система, находящаяся в равновесном состоянии, время от времени самопроизвольно отклоняется от
Система, находящаяся в равновесном состоянии, время от времени самопроизвольно отклоняется от







Термодинамической вероятностью или статисти-ческим весом макросостояния W − называется число макросостояний,
Термодинамической вероятностью или статисти-ческим весом макросостояния W − называется число макросостояний,

Вероятность сложного события, есть W =W1∙W2, где W1 – первое состояние;
Вероятность сложного события, есть W =W1∙W2, где W1 – первое состояние;

Например, в ящике черные и белые шары. Они порознь, есть порядок
Например, в ящике черные и белые шары. Они порознь, есть порядок

На этих рассуждениях Клаузиус в 1877 году и выдвинул гипотезу о
На этих рассуждениях Клаузиус в 1877 году и выдвинул гипотезу о

8. Третье начало термодинамики
Первое и Второе начало термодинамики не позволяет определить
8. Третье начало термодинамики
Первое и Второе начало термодинамики не позволяет определить

Обычно его формулируют следующим образом: энтропия любой равновесной системы при абсолютном
Обычно его формулируют следующим образом: энтропия любой равновесной системы при абсолютном

При T = 0, внутренняя энергия и тепловая функция системы прекращают
При T = 0, внутренняя энергия и тепловая функция системы прекращают

Третье начало термодинамики иногда формулируют следующим способом: при абсолютном нуле температуры
Третье начало термодинамики иногда формулируют следующим способом: при абсолютном нуле температуры

Следствием Третьего начала является, то что невозможно охладить тело до абсолютного
Следствием Третьего начала является, то что невозможно охладить тело до абсолютного

Лекция 29. Реальные газы
Реальные газы
Уравнение Ван-дер-Ваальса
Изотермы уравнения Ван-дер- Ваальса
Внутренняя энергия газа
Лекция 29. Реальные газы
Реальные газы
Уравнение Ван-дер-Ваальса
Изотермы уравнения Ван-дер- Ваальса
Внутренняя энергия газа

7.1. Реальные газы
Как известно, уравнение состояния устанавливает функциональную связь
7.1. Реальные газы
Как известно, уравнение состояния устанавливает функциональную связь

Самым простым и известным уравнением состояния является уравнение состояния идеального газа:
Самым простым и известным уравнением состояния является уравнение состояния идеального газа:

Для газов с низкой температурой сжиже-ния (He, H2, Ne и даже
Для газов с низкой температурой сжиже-ния (He, H2, Ne и даже

Наибольшее распространение вследствие простоты и физической наглядности получило уравнение Ван-дер-Ваальса
Наибольшее распространение вследствие простоты и физической наглядности получило уравнение Ван-дер-Ваальса

При понижении температуры межмолекулярное взаимодействие в реальных газах приводит к конденсации
При понижении температуры межмолекулярное взаимодействие в реальных газах приводит к конденсации

Ван-дер-Ваальс в 1873 г. Дал функциональную интерпретацию внутреннего давления. Согласно модели
Ван-дер-Ваальс в 1873 г. Дал функциональную интерпретацию внутреннего давления. Согласно модели

С учетом этих соображений уравнение состояния идеального газа преобразуется в уравнение
С учетом этих соображений уравнение состояния идеального газа преобразуется в уравнение

Помимо Нобелевской премии, Ван-дер-Ваальс получил почетную докторскую степень Кембриджского университета. Кроме
Помимо Нобелевской премии, Ван-дер-Ваальс получил почетную докторскую степень Кембриджского университета. Кроме

Реальные газы – газы, свойства которых зависят от взаимодействия молекул. В
Реальные газы – газы, свойства которых зависят от взаимодействия молекул. В

Я.Д. Ван-дер-Ваальс для объяснения свойств реальных газов и жидкостей, предположил, что
Я.Д. Ван-дер-Ваальс для объяснения свойств реальных газов и жидкостей, предположил, что

Ориентационные силы действуют между полярными молекулами – молекулами, обладающими дипольными или
Ориентационные силы действуют между полярными молекулами – молекулами, обладающими дипольными или
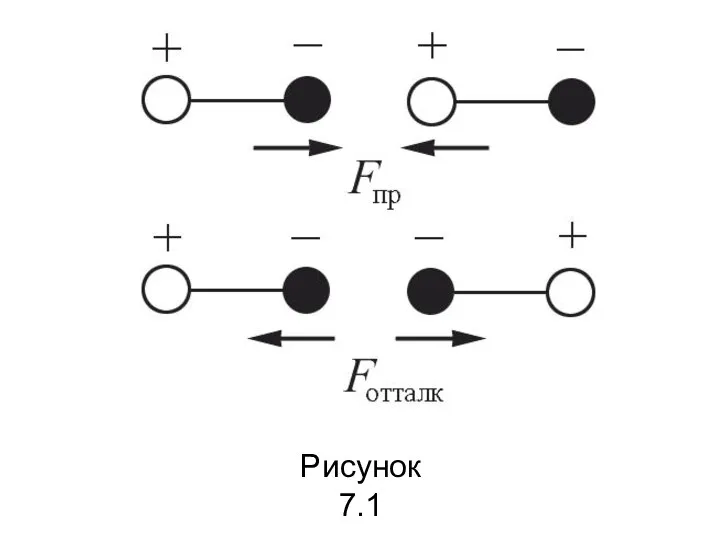
Рисунок 7.1
Рисунок 7.1

Среднее значение потенциальной энергии ориентационного межмолекулярного взаимодействия равно Uор(r) ~ Р1
Среднее значение потенциальной энергии ориентационного межмолекулярного взаимодействия равно Uор(r) ~ Р1

Индукционные (поляризационные) силы действуют между полярной и неполярной молекулами, а также
Индукционные (поляризационные) силы действуют между полярной и неполярной молекулами, а также

Индукционные силы убывают по тому же закону, что и ориентационные F
Индукционные силы убывают по тому же закону, что и ориентационные F

Данное взаимодействие называется дисперсионным, его энергия определяется поляризуемостью молекул α1, α2:
Данное взаимодействие называется дисперсионным, его энергия определяется поляризуемостью молекул α1, α2:

Отметим, что все три силы и энергии одинаковым образом убывают с
Отметим, что все три силы и энергии одинаковым образом убывают с

Силы отталкивания действуют между молекулами на очень малых расстояниях, когда происходит
Силы отталкивания действуют между молекулами на очень малых расстояниях, когда происходит

К хорошему согласию с данными экспериментов приводит допущение, что потенциальная энергия
К хорошему согласию с данными экспериментов приводит допущение, что потенциальная энергия
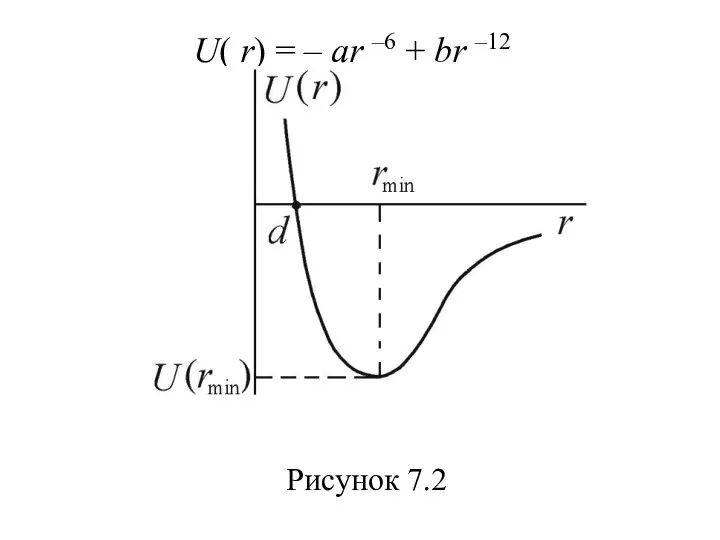
U( r) = – ar –6 + br –12
Рисунок 7.2
U( r) = – ar –6 + br –12
Рисунок 7.2

Глубина потенциала равна U(rmin) = –a2/4b при rmin = (2b/a)1/6 –
Глубина потенциала равна U(rmin) = –a2/4b при rmin = (2b/a)1/6 –

7.2. Уравнение Ван-дер-Ваальса
Уравнение Ван-дер-Ваальса – одно из первых уравнений
7.2. Уравнение Ван-дер-Ваальса
Уравнение Ван-дер-Ваальса – одно из первых уравнений

Учтем влияние конечных размеров молекул на уравнение состояния реального газа.
Учтем влияние конечных размеров молекул на уравнение состояния реального газа.

В результате в сосуде, содержащем N молекул конечных размеров, область объемом
В результате в сосуде, содержащем N молекул конечных размеров, область объемом

Объем, доступный точечным молекулам, будет равен V − b, а давление,
Объем, доступный точечным молекулам, будет равен V − b, а давление,

Для ν = m/μ молей газа уравнение состояния газа с учетом
Для ν = m/μ молей газа уравнение состояния газа с учетом

Рассмотрим теперь влияние сил притяжения на уравнение состояния идеального газа.
Рассмотрим теперь влияние сил притяжения на уравнение состояния идеального газа.

Это обусловлено тем, что в то время как в объеме газа
Это обусловлено тем, что в то время как в объеме газа
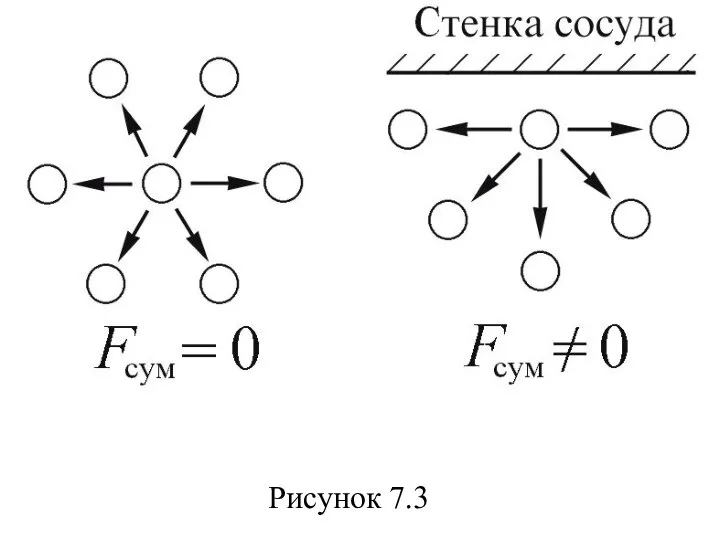
Рисунок 7.3
Рисунок 7.3

Дополнительное внутреннее давление пропорционально числу частиц, приходящихся на единицу площади границы
Дополнительное внутреннее давление пропорционально числу частиц, приходящихся на единицу площади границы

В результате избыточное внутреннее давление Pi (i − intrinsic) будет пропорционально
В результате избыточное внутреннее давление Pi (i − intrinsic) будет пропорционально

С учетом внутреннего давления уравнение состояния примет вид
P + Pi
С учетом внутреннего давления уравнение состояния примет вид P + Pi

Полученное уравнение с учетом выражения для Pi переходит в новое уравнение
Полученное уравнение с учетом выражения для Pi переходит в новое уравнение

Данное уравнение справедливо при условии νb << V и ν2a/V 2
Данное уравнение справедливо при условии νb << V и ν2a/V 2
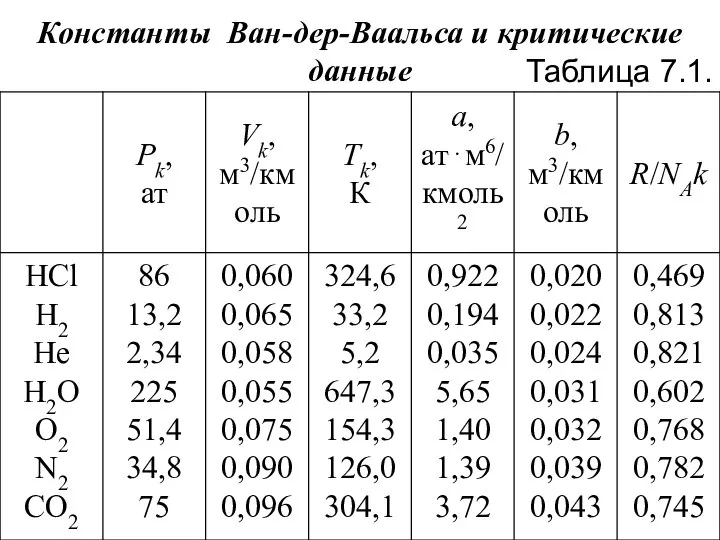
Константы Ван-дер-Ваальса и критические данные
Таблица 7.1.
Константы Ван-дер-Ваальса и критические данные
Таблица 7.1.

Примечание. Константы а и b выбраны таким образом, чтобы получить оптимальное
Примечание. Константы а и b выбраны таким образом, чтобы получить оптимальное

7.3. Изотермы уравнения Ван-дер-Ваальса
Проанализируем изотермы уравнения Ван-дер-Ваальса – зависимости Р от
7.3. Изотермы уравнения Ван-дер-Ваальса
Проанализируем изотермы уравнения Ван-дер-Ваальса – зависимости Р от

Поскольку данное уравнение имеет третью степень относительно V, а коэффициенты
Поскольку данное уравнение имеет третью степень относительно V, а коэффициенты
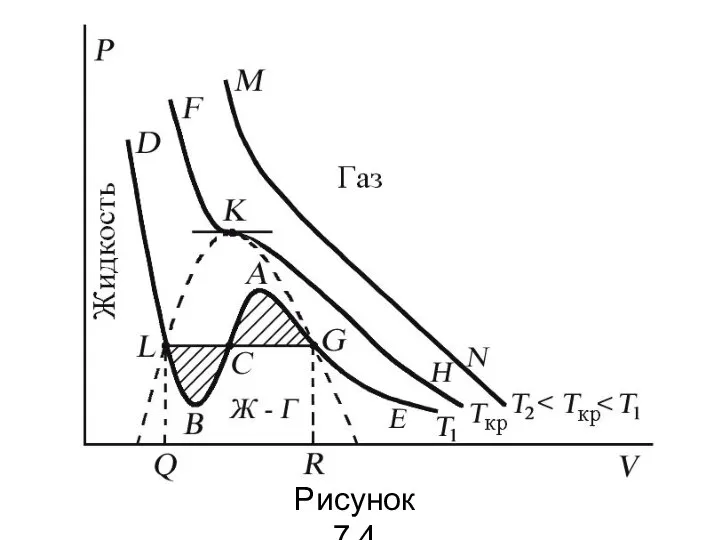
Рисунок 7.4
Рисунок 7.4

Изотерма при Ткр, которая разделяет немонотонные T < Tкр и
Изотерма при Ткр, которая разделяет немонотонные T < Tкр и

При температуре газа ниже критической такая однозначность исчезает, а это
При температуре газа ниже критической такая однозначность исчезает, а это
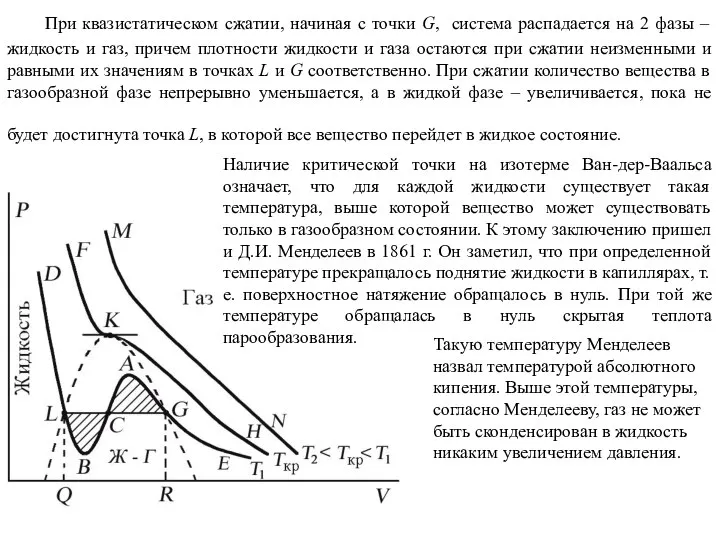
При квазистатическом сжатии, начиная с точки G, система распадается на
При квазистатическом сжатии, начиная с точки G, система распадается на

Критическую точку K мы определили как точку перегиба критической изотермы,
Критическую точку K мы определили как точку перегиба критической изотермы,

7.4. Внутренняя энергия газа Ван-дер-Ваальса
Энергия одного моля газа Ван-дер-Ваальса
7.4. Внутренняя энергия газа Ван-дер-Ваальса
Энергия одного моля газа Ван-дер-Ваальса

Потенциальная энергия притяжения молекул равна работе, необходимой для разведения молекул на
Потенциальная энергия притяжения молекул равна работе, необходимой для разведения молекул на

Знак «минус» указывает на то, что между молекулами действуют силы притяжения;
Знак «минус» указывает на то, что между молекулами действуют силы притяжения;

Принципиальное значение уравнения Ван-дер-Ваальса определяется следующими обстоятельствами:
1) уравнение было
Принципиальное значение уравнения Ван-дер-Ваальса определяется следующими обстоятельствами: 1) уравнение было

Причиной недостаточной точности уравнения Ван-дер-Ваальс считал ассоциацию молекул в газовой фазе,
Причиной недостаточной точности уравнения Ван-дер-Ваальс считал ассоциацию молекул в газовой фазе,
 Столкновения Абсолютно упругий удар
Столкновения Абсолютно упругий удар  Базовая электрика. Основы
Базовая электрика. Основы Тема 3: ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ 3.1. ЭНЕРГИЯ И ЭНТРОПИЯ Второй закон устанавливает, что самопроизвольные процессы возможн
Тема 3: ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ 3.1. ЭНЕРГИЯ И ЭНТРОПИЯ Второй закон устанавливает, что самопроизвольные процессы возможн Спектроскопия органических соединений
Спектроскопия органических соединений Соединение проводников
Соединение проводников Динамика судна. Общие понятия гидромеханики
Динамика судна. Общие понятия гидромеханики Прямолинейное и криволинейное движение. Движение тела по окружности
Прямолинейное и криволинейное движение. Движение тела по окружности Управління потоками реактивної енергії (вибір потужності компенсуючих установок та їх розміщення у системі електропостачання)
Управління потоками реактивної енергії (вибір потужності компенсуючих установок та їх розміщення у системі електропостачання) « МОУ Липковская Средняя Школа №3» Презентация на тему: « Диффузия» ПОДГОТОВИЛИ: НИКИТИНА НАДЕЖДА И ХАЛИМОВА ЕКАТЕРИНА
« МОУ Липковская Средняя Школа №3» Презентация на тему: « Диффузия» ПОДГОТОВИЛИ: НИКИТИНА НАДЕЖДА И ХАЛИМОВА ЕКАТЕРИНА Электр тізбектеріндегі өтпелі үрдістер
Электр тізбектеріндегі өтпелі үрдістер Законы постоянного тока
Законы постоянного тока Плавление тел
Плавление тел Сила трения
Сила трения Лазер. Области применения лазеров
Лазер. Области применения лазеров Arduino. Электрическая схема
Arduino. Электрическая схема Задачи по физике
Задачи по физике харчування під час радіоактивної небезпеки Підготувала учениця 302 групи Авіакосмічного ліцею ім.І.Сікорського НАУ Гріднєва А
харчування під час радіоактивної небезпеки Підготувала учениця 302 групи Авіакосмічного ліцею ім.І.Сікорського НАУ Гріднєва А Методы травления материалов электронной техники
Методы травления материалов электронной техники Электрические цепи постоянного тока
Электрические цепи постоянного тока Энергетические ресурсы Мирового океана
Энергетические ресурсы Мирового океана Макс Планк Презентация по физике ученицы 12а класса Рижской Гризинькалнской средней шк. им. И. Г. Гердера Максимовой Татьяны
Макс Планк Презентация по физике ученицы 12а класса Рижской Гризинькалнской средней шк. им. И. Г. Гердера Максимовой Татьяны  Уравнение адиабатического процесса для идеального газа
Уравнение адиабатического процесса для идеального газа Закон Архимеда
Закон Архимеда Турбины ТЭС и АЭС. Теория теплового процесса. Основные уравнения теории
Турбины ТЭС и АЭС. Теория теплового процесса. Основные уравнения теории Вес тела
Вес тела Парообразование. Конденсация
Парообразование. Конденсация Презентация по физике "Физика-наука о природе" - скачать
Презентация по физике "Физика-наука о природе" - скачать  Презентация по физике на тему «Производство электроэнергии. Передача электроэнергии на расстоянии.» Ученицы 9 класса «В» Зениной
Презентация по физике на тему «Производство электроэнергии. Передача электроэнергии на расстоянии.» Ученицы 9 класса «В» Зениной