- Next-generation sequencing RNA-Seq. Анализ

Содержание
- 2. Шаг 5: Анализ данных
- 3. Особенности программного обеспечения для анализа данных NGS Анализ данных NGS. Брагин Антон, Sequoia Genetics. 2013. Модульная
- 4. ДНК A Приготовление библиотеки B Секвенирование D Анализ данных
- 5. Анализ данных Ion Torrent Определение положений на чипе, в которых находятся последовательности Перевод последовательности сигналов в
- 6. Ошибки случаются… Эксперимент. Ошибки постановки эксперимента Биоинформатика. Ошибки в чтениях возникают из-за неточности работы секвенатора Контроль
- 7. Контроль и улучшение качества результатов секвенирования Ошибки Сырые данные собираются в DAT файлы на Ion PGM
- 8. 1) Димеры адаптеров. Адаптеры соединяются друг с другом, без фрагмента ДНК образца Возможные ошибки Barry Merriman,
- 9. Возможные ошибки Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress in Ion Torrent semiconductor
- 10. Тримминг - удаление ошибок секвенирования Две задачи тримминга: 1. Удаление последовательности адаптера в чтениях 2. Отсечение
- 11. P – вероятность ошибки Q – параметр качества (Phred Quality Score) FASTQ – стандартный формат записи
- 12. • Синяя линия – среднее качество • Красная линия – медиана • Жёлтая рамка – интерквартиль
- 13. FASTQ – общепринятый формат записи чтений. FASTQ – стандартный формат записи чтений Barry Merriman, Ion Torrent
- 14. Качество в формате FASTQ закодировано в ASCII – символах Существовало несколько стандартов записи качества Многим программам
- 15. Первичный анализ Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H. S., & Morling, N.
- 16. Первичный анализ Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H. S., & Morling, N.
- 17. Фильтры для ридов Варианты фильтрации прочтений: 1. Удаление коротких прочтений 2. Удаление димеров адаптеров 3. Удаление
- 18. Фильтр на удаление поликлональных прочтений Technical Note. Trimming and Filtering http://mendel.iontorrent.com/ion-docs/Technical-Note---Filtering-and-Trimming_6455370.html
- 19. Torrent Suite™ Data Analysis Flow
- 20. Example shown for 100bp (260 flow) run on Ion PITM Chip (TAH-191) Ion Proton™ System Pipeline
- 21. Example shown for 100bp (260 flow) run on Ion PITM Chip (TAH-191) Ion Proton™ System Pipeline
- 22. Вторичный анализ (Обработка BAM файлов при помощи плагинов) Methods, tools, and pipelines for analysis of Ion
- 23. Шаг 1: Тримминг по качеству 3’ конца и Ion P1B адаптеру Шаг 2: Оценка и контроль
- 24. Анализ транскриптома. Рабочий процесс Life Technologies—Sample to RNA-Seq. 2012 Обсчет/статистика Картирование Оценка качества Предварительная обработка данных
- 25. Ion Proton™ System Enables High Quality Transcriptome Analysis with >80M Reads per Run Ion Proton™ Runs
- 26. Дополнительный шаг по обрезанию концов. Низкое качество и фрагменты адаптерной последовательности приводят к ошибкам картирования или
- 27. Программа FastQC использует FASTQ файл в качестве входных данных Качество прочтений по каждому нуклеотиду Оценка качества
- 28. Ошибки можно не только удалять, но и исправлять Коррекция чтений Программы, исправляющие ошибки, основаны на подсчёте
- 29. Что такое kmer size? Как собрать геном de novo из коротких чтений? Практические советы. Науменко С.А.
- 30. Для анализа транскриптома следует использовать параметр с длиной фрагмента для анализа 18 нуклеотидов (и количество допустимых
- 31. SAM-формат (Sequence Alignment/Map format) – текстовый формат, предназначенный для представления информация о картировании чтений. Значения отдельных
- 32. BAM-формат (Binary Sequence Alignment/Map) - Сжатый бинарный вариант формата SAM. Для быстрого доступа к данным по
- 33. Integrative Genomics Viewer – программа для визуального просмотра BAM-файлов
- 34. Ion Torrent data from RNA-Seq analysis of a Ewings Sarcoma cell line (Data courtesy T. Triche,
- 35. Повтор Прочтения YIPF2 с антисмысловой цепи Структура гена YIPF2 Структура гена CARM1 Прочтения, соответствующие транскрипту CARM1
- 36. EWSR1/FLI1 fusion protein type 1 (EWSR1/FLI1 fusion) mRNA Ion Torrent Internal Data Ion Torrent data from
- 37. Шаг 4: Подсчет картированных прочтений Скрипт htseq-count.py для подсчета картированных прочтений Скрипт – простая программа, предназначенная
- 38. Spike-in РНК: известна последовательность известна конечная концентрация используется для оценки точности измерений дифференциальной экспрессии генов 92
- 39. Контроль ERCC RNA Spike-In Control Mixes Характеристики библиотеки высокого качества: R2 около 0.9 Размер выборки –
- 40. TaqMan Expression Data with MAQC Ion Proton™ Expression Data with MAQC R2 = 0.958 N= 657
- 41. Высокий коэффициент корреляции с данными микрочипов Усредненные данные трех чипов 314 на образец РНК сравнивались с
- 42. Скрипт на Perl анализирует SAM файл на базовую статистику по картированию: общее количество картированных ридов, риды,
- 43. Нормализация данных RNA-Seq необходима из-за различий в глубине секвенирования, длине генов, отличий между образцами по количеству
- 44. Обобщенные статистические подходы к анализу транскриптомных данных и рекомендации по данному вопросу описаны в Current Protocols
- 45. Анализ микроРНК. Рабочий процесс Life Technologies—Sample to RNA-Seq. 2012 Обсчет/статистика Картирование Оценка качества Предварительная обработка данных
- 46. FASTX-toolkit – программный пакет набора инструментов для обработки и оценки FASTQ файлов. fastq_quality_trimmer применяется таким же
- 47. Первый шаг картирования – шпильки микроРНК. Предшественники микроРНК (60-90 нуклеотидов) хранятся в базе RFAM miRBase database
- 48. Шаг 4: Подсчет картированных прочтений Скрипт htseq-count.py для подсчета картированных прочтений Что делает: из SAM-файла извлекает
- 49. Скрипт на Perl анализирует SAM файл на базовую статистику по картированию: общее количество картированных ридов, риды,
- 50. Torrent Server Torrent Browser Оценка качества в программе Torrent suite Специальные модули для разных приложений Анализ
- 51. Облако Biological Interpretation Partek® Genomics Suite™ Partek® Flow™ Partek® Pathway™ Сырые данные Copyright © 2011 Partek
- 52. Характеристики Интеграция с Torrent Suite™ Software Работает в облаке и на кластере Online доступ к данным
- 53. Количественное картирование транскриптома Обнаружение новых транскриптов Использование всех доступных баз данных Оценка представленности отдельных изоформ Анализ
- 55. Скачать презентацию

Шаг 5: Анализ данных
Шаг 5: Анализ данных

Особенности программного обеспечения
для анализа данных NGS
Анализ данных NGS. Брагин Антон,
Особенности программного обеспечения
для анализа данных NGS
Анализ данных NGS. Брагин Антон,

ДНК
A
Приготовление
библиотеки
B
Секвенирование
D
Анализ данных
ДНК
A
Приготовление
библиотеки
B
Секвенирование
D
Анализ данных

Анализ данных Ion Torrent
Определение положений на чипе, в которых находятся последовательности
Перевод
Анализ данных Ion Torrent
Определение положений на чипе, в которых находятся последовательности
Перевод

Ошибки случаются…
Эксперимент. Ошибки постановки эксперимента
Биоинформатика. Ошибки в чтениях возникают из-за неточности
Ошибки случаются…
Эксперимент. Ошибки постановки эксперимента
Биоинформатика. Ошибки в чтениях возникают из-за неточности
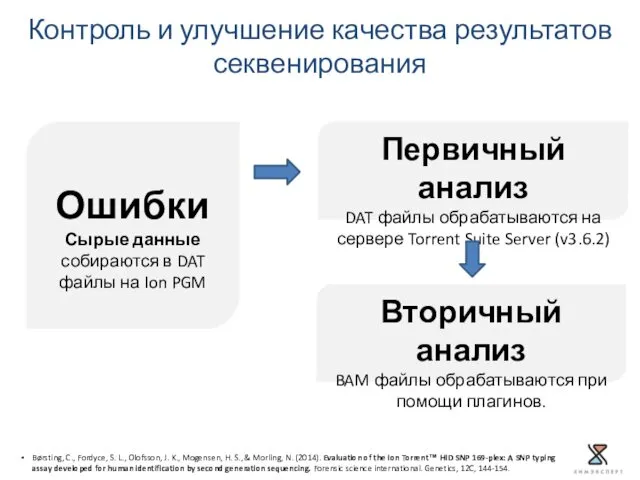
Контроль и улучшение качества результатов
секвенирования
Ошибки Сырые данные собираются в DAT файлы
Контроль и улучшение качества результатов
секвенирования
Ошибки Сырые данные собираются в DAT файлы

1) Димеры адаптеров. Адаптеры соединяются друг с другом, без фрагмента ДНК
1) Димеры адаптеров. Адаптеры соединяются друг с другом, без фрагмента ДНК

Возможные ошибки
Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress
Возможные ошибки
Barry Merriman, Ion Torrent R&D Team, Jonathan M. Rothberg. Progress

Тримминг - удаление ошибок секвенирования
Две задачи тримминга:
1. Удаление последовательности адаптера в
Тримминг - удаление ошибок секвенирования
Две задачи тримминга:
1. Удаление последовательности адаптера в

P – вероятность ошибки
Q – параметр качества (Phred Quality Score)
FASTQ –
P – вероятность ошибки
Q – параметр качества (Phred Quality Score)
FASTQ –
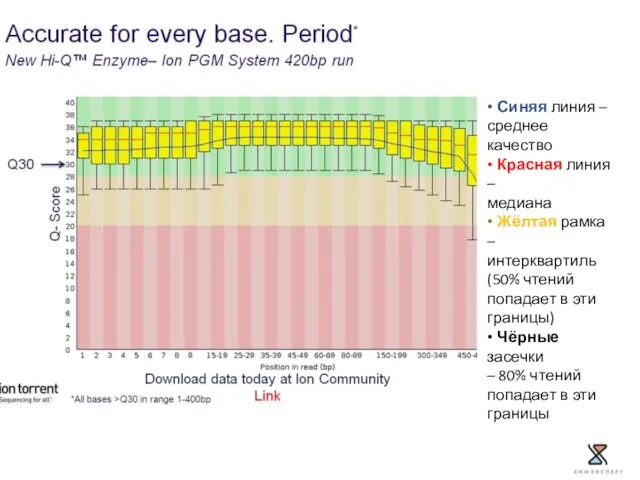
• Синяя линия –
среднее качество
• Красная линия –
медиана
• Жёлтая рамка –
интерквартиль
(50%
• Синяя линия –
среднее качество
• Красная линия –
медиана
• Жёлтая рамка –
интерквартиль
(50%

FASTQ – общепринятый формат записи чтений.
FASTQ – стандартный формат
записи чтений
Barry Merriman,
FASTQ – общепринятый формат записи чтений.
FASTQ – стандартный формат
записи чтений
Barry Merriman,

Качество в формате FASTQ закодировано в ASCII – символах
Существовало несколько стандартов
Качество в формате FASTQ закодировано в ASCII – символах
Существовало несколько стандартов

Первичный анализ
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H.
Первичный анализ
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H.

Первичный анализ
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H.
Первичный анализ
Børsting, C., Fordyce, S. L., Olofsson, J. K., Mogensen, H.

Фильтры для ридов
Варианты фильтрации прочтений:
1. Удаление коротких прочтений
2. Удаление димеров адаптеров
3.
Фильтры для ридов
Варианты фильтрации прочтений:
1. Удаление коротких прочтений
2. Удаление димеров адаптеров
3.
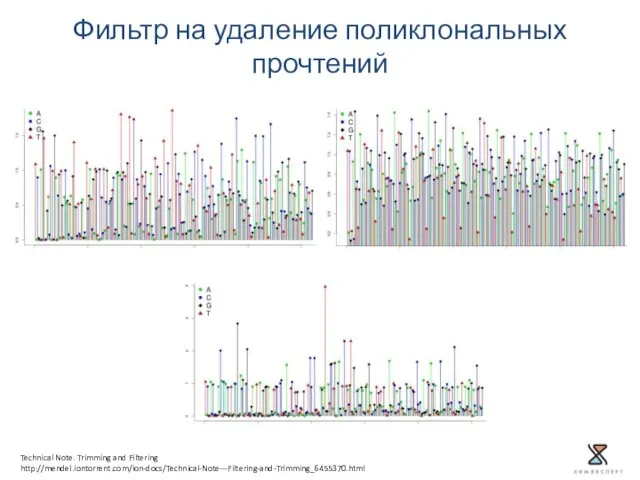
Фильтр на удаление поликлональных прочтений
Technical Note. Trimming and Filtering
http://mendel.iontorrent.com/ion-docs/Technical-Note---Filtering-and-Trimming_6455370.html
Фильтр на удаление поликлональных прочтений
Technical Note. Trimming and Filtering
http://mendel.iontorrent.com/ion-docs/Technical-Note---Filtering-and-Trimming_6455370.html

Torrent Suite™ Data Analysis Flow
Torrent Suite™ Data Analysis Flow

Example shown for 100bp (260 flow) run on Ion PITM Chip
Example shown for 100bp (260 flow) run on Ion PITM Chip

Example shown for 100bp (260 flow) run on Ion PITM Chip
Example shown for 100bp (260 flow) run on Ion PITM Chip

Вторичный анализ
(Обработка BAM файлов при помощи плагинов)
Methods, tools, and pipelines for
Вторичный анализ
(Обработка BAM файлов при помощи плагинов)
Methods, tools, and pipelines for

Шаг 1: Тримминг по качеству 3’ конца и Ion P1B адаптеру
Шаг
Шаг 1: Тримминг по качеству 3’ конца и Ion P1B адаптеру
Шаг

Анализ транскриптома. Рабочий процесс
Life Technologies—Sample to RNA-Seq. 2012
Обсчет/статистика
Картирование
Оценка качества
Предварительная обработка данных
Анализ транскриптома. Рабочий процесс
Life Technologies—Sample to RNA-Seq. 2012
Обсчет/статистика
Картирование
Оценка качества
Предварительная обработка данных

Ion Proton™ System Enables High Quality Transcriptome Analysis with >80M Reads
Ion Proton™ System Enables High Quality Transcriptome Analysis with >80M Reads

Дополнительный шаг по обрезанию концов.
Низкое качество и фрагменты адаптерной последовательности приводят
Дополнительный шаг по обрезанию концов.
Низкое качество и фрагменты адаптерной последовательности приводят

Программа FastQC использует FASTQ файл в качестве входных данных
Качество прочтений по
Программа FastQC использует FASTQ файл в качестве входных данных
Качество прочтений по

Ошибки можно не только удалять, но и исправлять
Коррекция чтений
Программы, исправляющие ошибки,
Ошибки можно не только удалять, но и исправлять
Коррекция чтений
Программы, исправляющие ошибки,
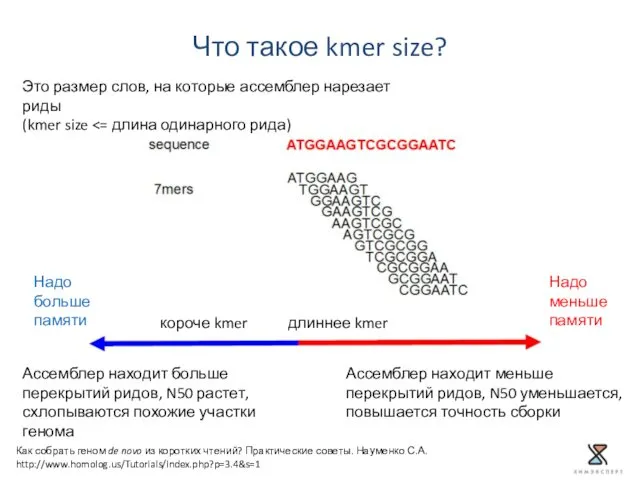
Что такое kmer size?
Как собрать геном de novo из коротких чтений?
Что такое kmer size?
Как собрать геном de novo из коротких чтений?

Для анализа транскриптома следует использовать параметр с длиной фрагмента для анализа
Для анализа транскриптома следует использовать параметр с длиной фрагмента для анализа

SAM-формат
(Sequence Alignment/Map format)
– текстовый формат, предназначенный для представления информация о
SAM-формат
(Sequence Alignment/Map format)
– текстовый формат, предназначенный для представления информация о

BAM-формат
(Binary Sequence Alignment/Map)
- Сжатый бинарный вариант формата SAM. Для быстрого
BAM-формат
(Binary Sequence Alignment/Map)
- Сжатый бинарный вариант формата SAM. Для быстрого
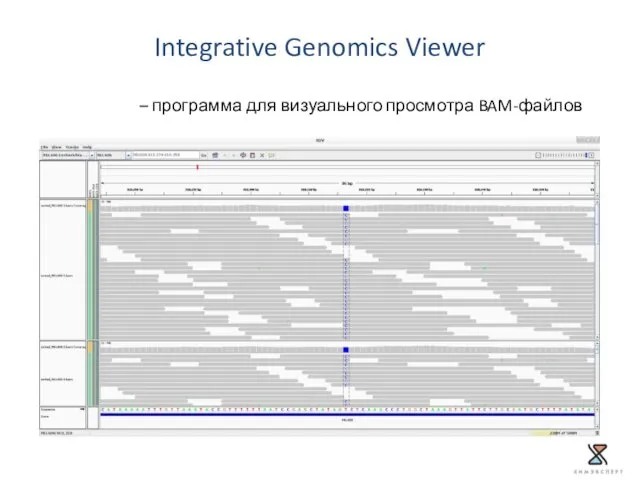
Integrative Genomics Viewer
– программа для визуального просмотра BAM-файлов
Integrative Genomics Viewer
– программа для визуального просмотра BAM-файлов
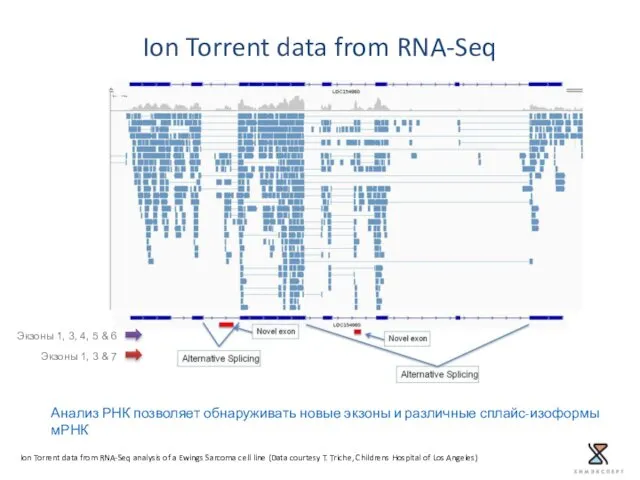
Ion Torrent data from RNA-Seq analysis of a Ewings Sarcoma cell
Ion Torrent data from RNA-Seq analysis of a Ewings Sarcoma cell
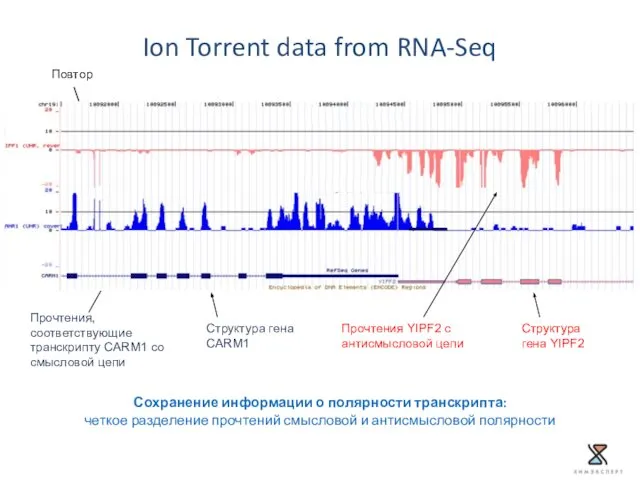
Повтор
Прочтения YIPF2 с антисмысловой цепи
Структура гена YIPF2
Структура гена CARM1
Прочтения, соответствующие
Повтор
Прочтения YIPF2 с антисмысловой цепи
Структура гена YIPF2
Структура гена CARM1
Прочтения, соответствующие

EWSR1/FLI1 fusion protein type 1 (EWSR1/FLI1 fusion) mRNA
Ion Torrent Internal Data
Ion
EWSR1/FLI1 fusion protein type 1 (EWSR1/FLI1 fusion) mRNA
Ion Torrent Internal Data
Ion

Шаг 4: Подсчет картированных прочтений
Скрипт htseq-count.py для подсчета картированных прочтений
Скрипт –
Шаг 4: Подсчет картированных прочтений
Скрипт htseq-count.py для подсчета картированных прочтений
Скрипт –

Spike-in РНК:
известна последовательность
известна конечная концентрация
используется для оценки точности измерений дифференциальной
Spike-in РНК:
известна последовательность
известна конечная концентрация
используется для оценки точности измерений дифференциальной
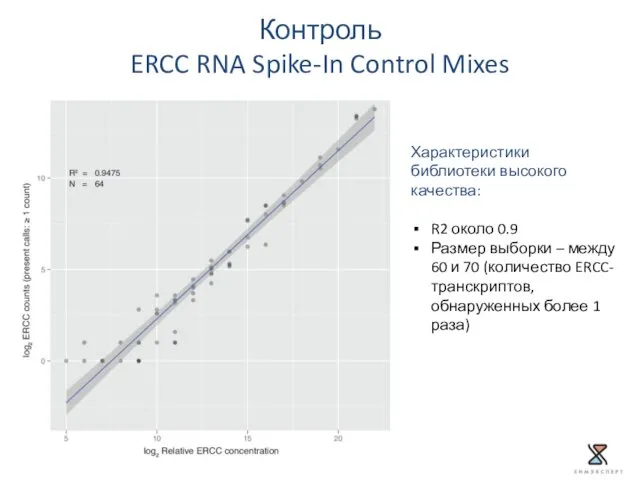
Контроль
ERCC RNA Spike-In Control Mixes
Характеристики библиотеки высокого качества:
R2 около 0.9
Размер
Контроль
ERCC RNA Spike-In Control Mixes
Характеристики библиотеки высокого качества:
R2 около 0.9
Размер

TaqMan Expression Data with MAQC
Ion Proton™ Expression Data with MAQC
R2 =
TaqMan Expression Data with MAQC
Ion Proton™ Expression Data with MAQC
R2 =

Высокий коэффициент корреляции с данными микрочипов
Усредненные данные трех чипов 314 на
Высокий коэффициент корреляции с данными микрочипов
Усредненные данные трех чипов 314 на

Скрипт на Perl анализирует SAM файл на базовую статистику по картированию:
Скрипт на Perl анализирует SAM файл на базовую статистику по картированию:

Нормализация данных RNA-Seq необходима из-за различий в
глубине секвенирования,
длине генов,
Нормализация данных RNA-Seq необходима из-за различий в
глубине секвенирования,
длине генов,

Обобщенные статистические подходы к анализу транскриптомных данных и рекомендации по данному
Обобщенные статистические подходы к анализу транскриптомных данных и рекомендации по данному
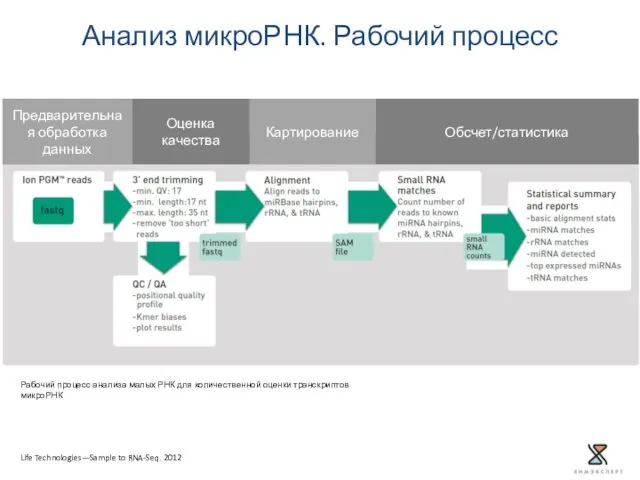
Анализ микроРНК. Рабочий процесс
Life Technologies—Sample to RNA-Seq. 2012
Обсчет/статистика
Картирование
Оценка качества
Предварительная обработка данных
Рабочий
Анализ микроРНК. Рабочий процесс
Life Technologies—Sample to RNA-Seq. 2012
Обсчет/статистика
Картирование
Оценка качества
Предварительная обработка данных
Рабочий

FASTX-toolkit – программный пакет набора инструментов для обработки и оценки FASTQ
FASTX-toolkit – программный пакет набора инструментов для обработки и оценки FASTQ

Первый шаг картирования – шпильки микроРНК.
Предшественники микроРНК (60-90 нуклеотидов) хранятся
Первый шаг картирования – шпильки микроРНК.
Предшественники микроРНК (60-90 нуклеотидов) хранятся

Шаг 4: Подсчет картированных прочтений
Скрипт htseq-count.py для подсчета картированных прочтений
Что делает:
из
Шаг 4: Подсчет картированных прочтений
Скрипт htseq-count.py для подсчета картированных прочтений
Что делает:
из

Скрипт на Perl анализирует SAM файл на базовую статистику по картированию:
Скрипт на Perl анализирует SAM файл на базовую статистику по картированию:
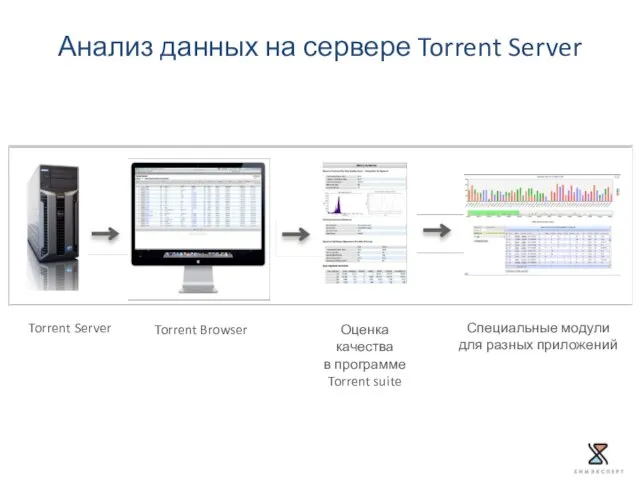
Torrent Server
Torrent Browser
Оценка качества
в программе
Torrent suite
Специальные модули
для разных приложений
Анализ
Torrent Server
Torrent Browser
Оценка качества
в программе
Torrent suite
Специальные модули
для разных приложений
Анализ

Облако
Biological
Interpretation
Partek® Genomics Suite™
Partek® Flow™
Partek®
Pathway™
Сырые данные
Copyright © 2011 Partek Incorporated. All
Облако
Biological
Interpretation
Partek® Genomics Suite™
Partek® Flow™
Partek®
Pathway™
Сырые данные
Copyright © 2011 Partek Incorporated. All

Характеристики
Интеграция с Torrent Suite™ Software
Работает в облаке и на кластере
Online доступ
Характеристики
Интеграция с Torrent Suite™ Software
Работает в облаке и на кластере
Online доступ

Количественное картирование транскриптома
Обнаружение новых транскриптов
Использование всех доступных баз данных
Оценка представленности
Количественное картирование транскриптома
Обнаружение новых транскриптов
Использование всех доступных баз данных
Оценка представленности
 Локальные вычислительные сети и их виды. Локальные вычислительные сети и их виды. Основные требования, предъявляемые к локал
Локальные вычислительные сети и их виды. Локальные вычислительные сети и их виды. Основные требования, предъявляемые к локал Устройство компьтера
Устройство компьтера Проектная деятельность на уроках информатики
Проектная деятельность на уроках информатики Архитектура, принципы построения современных вычислительных систем и комплексов. Вычислительные комплексы и сети систем РКО
Архитектура, принципы построения современных вычислительных систем и комплексов. Вычислительные комплексы и сети систем РКО АЛГОРИТМЫ В НАШЕЙ ЖИЗНИ урок информатики в 5 классе Учитель математики : Смолина Т.Г.
АЛГОРИТМЫ В НАШЕЙ ЖИЗНИ урок информатики в 5 классе Учитель математики : Смолина Т.Г. Қолданбалы программалар пакеті. Программалық жабдықтар
Қолданбалы программалар пакеті. Программалық жабдықтар Электронные таблицы Microsoft Excel
Электронные таблицы Microsoft Excel Инструменты и методы разработки интернет-проектов
Инструменты и методы разработки интернет-проектов Автоматизация комплекса задач менеджера отдела логистики ООО Эпсилон на платформе MS Access
Автоматизация комплекса задач менеджера отдела логистики ООО Эпсилон на платформе MS Access Занимательные задачи информатики. Задачи о лжецах
Занимательные задачи информатики. Задачи о лжецах Як ми виграли комп‘ютер. Гра-тест
Як ми виграли комп‘ютер. Гра-тест ИСПОЛЬЗОВАНИЕ СОВРЕМЕННЫХ ИНФОРМАЦИОННЫХ И КОММУНИКАЦИОННЫХ ТЕХНОЛОГИЙ В ОБРАЗОВАНИИ Презентация учебной дисциплины Москва -200
ИСПОЛЬЗОВАНИЕ СОВРЕМЕННЫХ ИНФОРМАЦИОННЫХ И КОММУНИКАЦИОННЫХ ТЕХНОЛОГИЙ В ОБРАЗОВАНИИ Презентация учебной дисциплины Москва -200 Кодирование
Кодирование Как делать анимацию
Как делать анимацию СРС-2
СРС-2 Blind date
Blind date Презентация "креативные индустрии" - скачать презентации по Информатике
Презентация "креативные индустрии" - скачать презентации по Информатике Автоматизированные системы, интенсивно использующие ПО
Автоматизированные системы, интенсивно использующие ПО Процессы и задачи. Операционные системы. Лекция 2
Процессы и задачи. Операционные системы. Лекция 2 Использование версии 30 ПИ Судебное делопроизводство ГАС Правосудие
Использование версии 30 ПИ Судебное делопроизводство ГАС Правосудие Команды mmx/xmm
Команды mmx/xmm Компьютерные сети
Компьютерные сети Кодирование текстовой информации
Кодирование текстовой информации MagicDiagram.com
MagicDiagram.com Информатика в играх и задачах. Основы логики. 2 класс ( 1 урок)
Информатика в играх и задачах. Основы логики. 2 класс ( 1 урок) Алгоритмизация и программирование. Понятие, свойства и способы записи алгоритмов. (Тема 5)
Алгоритмизация и программирование. Понятие, свойства и способы записи алгоритмов. (Тема 5) Проектирование баз данных
Проектирование баз данных Среда программирования Scratch. Занятие 3
Среда программирования Scratch. Занятие 3