- Болезнь Тея-Сакса

Содержание
- 2. Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) —редкое наследственное заболевание с аутосомно-рецессивным типом
- 3. Тей Уоррен (1843-1927) Сакс Теодор Бернард (1858-1944)
- 4. Гексозаминидаза А (НЕХА) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида — гексозаминидазы. Он способствует расщеплению жировых
- 5. Различают три формы болезни Тея - Сакса: 1. Детская форма — через полгода после рождения у
- 6. 2. Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания)дизартрия,(расстройства речи), атаксия (шаткость походки), спастичность (контрактуры
- 7. Болезнь распространена у евреев-ашкеназов. Среди них около 3 % являются носителями мутации в гене HEXA. Также
- 8. Клиническая картина: 1. Хронический дефицит гексозаминидазы типа А: Новорожденные в первые месяцы жизни развиваются нормально. Однако,
- 9. 2. Ювенильный дефицит гексозаминидазы типа А. Период начала заболевания с 14 до 30 лет. У взрослых
- 10. Диагностика. Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения специалист обычно может обнаружить на
- 11. Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях и тканях исследуемого. Необходимы анализ
- 12. В диагностике заболевания важнейшая роль принадлежит генетическому анализу. Также проводят анализ крови для определения уровня гексозаминидазы
- 13. Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез — анализ амниотической жидкости, полученной при проколе
- 15. Болезнь Тея—Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и также постепенно ведет к угасанию:
- 17. Скачать презентацию

Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) —редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную
Боле́знь Тея — Са́кса (GM2 ганглиозидоз, ранняя детская амавротическая идиотия) —редкое наследственное заболевание с аутосомно-рецессивным типом наследования, поражающее центральную

Тей Уоррен
(1843-1927)
Сакс Теодор Бернард (1858-1944)
Тей Уоррен
(1843-1927)
Сакс Теодор Бернард (1858-1944)

Гексозаминидаза А (НЕХА) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида —
Гексозаминидаза А (НЕХА) - лизосомный фермент, катализирующий катаболизм GM2 ганглиозида —

Различают три формы болезни
Тея - Сакса:
1. Детская форма — через полгода
Различают три формы болезни
Тея - Сакса:
1. Детская форма — через полгода

2. Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания)дизартрия,(расстройства речи), атаксия
(шаткость походки), спастичность (контрактуры и параличи). Смерть
2. Подростковая форма — развиваются моторно-когнитивные проблемы, дисфагия (нарушение глотания)дизартрия,(расстройства речи), атаксия
(шаткость походки), спастичность (контрактуры и параличи). Смерть

Болезнь распространена у евреев-ашкеназов. Среди них около 3 % являются носителями мутации
Болезнь распространена у евреев-ашкеназов. Среди них около 3 % являются носителями мутации

Клиническая картина:
1. Хронический дефицит гексозаминидазы типа А: Новорожденные в первые месяцы жизни
Клиническая картина:
1. Хронический дефицит гексозаминидазы типа А: Новорожденные в первые месяцы жизни

2. Ювенильный дефицит гексозаминидазы типа А. Период начала заболевания с 14
2. Ювенильный дефицит гексозаминидазы типа А. Период начала заболевания с 14
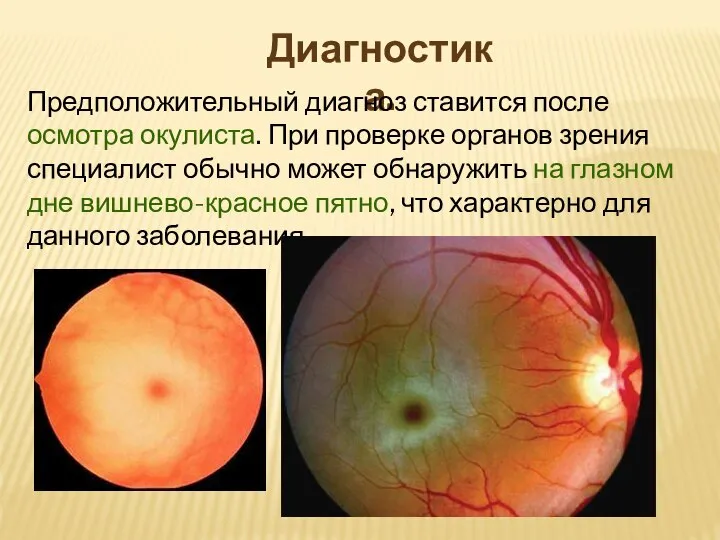
Диагностика.
Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения
Диагностика.
Предположительный диагноз ставится после осмотра окулиста. При проверке органов зрения
Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях
Далее подтвердить предположения помогает анализ на определение количества фермента в жидкостях

В диагностике заболевания важнейшая роль принадлежит генетическому анализу.
Также проводят анализ крови
В диагностике заболевания важнейшая роль принадлежит генетическому анализу.
Также проводят анализ крови
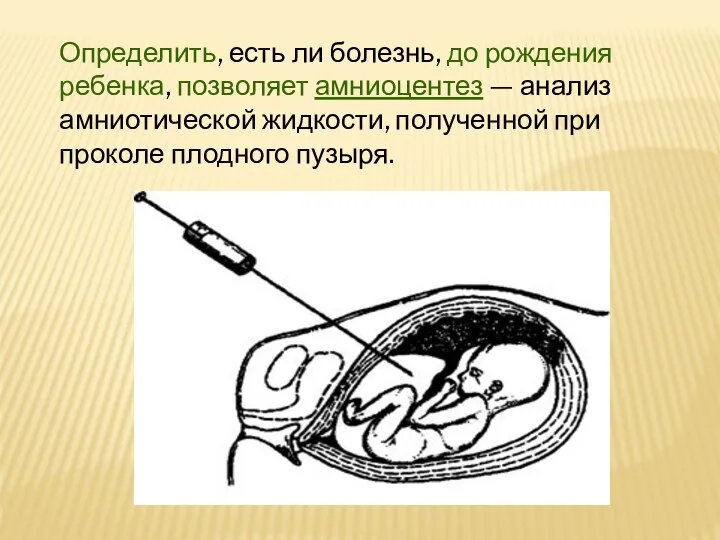
Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез — анализ
Определить, есть ли болезнь, до рождения ребенка, позволяет амниоцентез — анализ


Болезнь Тея—Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и
Болезнь Тея—Сакса не поддается лечению. Клиническая картина обычно нарастает постепенно и
 Облитерирующий атеросклероз брюшной аорты и артерий нижних конечностей
Облитерирующий атеросклероз брюшной аорты и артерий нижних конечностей Причины травматизма в школьном возрасте и пути их предотвращения
Причины травматизма в школьном возрасте и пути их предотвращения Определение доказательной медицины. История развития доказательной медицины. Мировой опыт развития
Определение доказательной медицины. История развития доказательной медицины. Мировой опыт развития Период молочных зубов. Преддошкольный и дошкольный периоды
Период молочных зубов. Преддошкольный и дошкольный периоды Введение прикормов
Введение прикормов Ингибиторы протеаз
Ингибиторы протеаз Мягкие лекарственные формы
Мягкие лекарственные формы Внутриутробная инфекция плода и новорожденного
Внутриутробная инфекция плода и новорожденного Imobilitātes radītās kaitīgās sekas cilvēka organismā –izmaiņas ķermeņa sistēmās
Imobilitātes radītās kaitīgās sekas cilvēka organismā –izmaiņas ķermeņa sistēmās Gem_8-2022
Gem_8-2022 Градація та узагальнення доказів
Градація та узагальнення доказів Тазовое предлежание
Тазовое предлежание Неспецифические воспалительные заболевания органов мочевыводящих путей
Неспецифические воспалительные заболевания органов мочевыводящих путей Энурез. Эпидемиология
Энурез. Эпидемиология Науқастың функсиональды жағдайын бағалау тыныс алуын, пульсін есептеу,артериялық қысымын өлшеу дағдыларын өз бетінше дамыту
Науқастың функсиональды жағдайын бағалау тыныс алуын, пульсін есептеу,артериялық қысымын өлшеу дағдыларын өз бетінше дамыту Классификация, клиника диагностика и лечение болезни Паркинсона
Классификация, клиника диагностика и лечение болезни Паркинсона День медицинского работника. Медицинский персонал Ягодинской больницы
День медицинского работника. Медицинский персонал Ягодинской больницы Этапы становления речи детей (по А.Н. Леонтьеву)
Этапы становления речи детей (по А.Н. Леонтьеву) Воспалительные заболевания женских половых органов
Воспалительные заболевания женских половых органов Скажем вирусу стоп
Скажем вирусу стоп Major public health issues in Sri Lanka: recovery of the post-conflict health system in north east Sri Lanka
Major public health issues in Sri Lanka: recovery of the post-conflict health system in north east Sri Lanka Общая рецептура
Общая рецептура Психотерапия. Психотерапевтический подход (неспецифическая психотерапия)
Психотерапия. Психотерапевтический подход (неспецифическая психотерапия) Хвороби органів дихання та їх профілактика
Хвороби органів дихання та їх профілактика Адгезия. Условия адгезии. Стандартизация стоматологических материалов
Адгезия. Условия адгезии. Стандартизация стоматологических материалов Physiology of Pregnancy
Physiology of Pregnancy Безопасность здоровья. Профилактика коронавирусной инфекции
Безопасность здоровья. Профилактика коронавирусной инфекции Травма. Виды травм
Травма. Виды травм