- Гипопластические состояния кроветворения. Классификация, патогенез, клиника, диагностика и лечение

Содержание
- 2. ПРИЧИНЫ ЦИТОПЕНИЧЕСКИХ СОСТОЯНИЙ НАРУШЕНИЕ ПРОДУКЦИИ КЛЕТОК КРОВИ ПОВЫШЕННОЕ РАЗРУШЕНИЕ КЛЕТОК КРОВИ ПЕРЕРАСПРЕДЕЛЕНИЕ КЛЕТОК КРОВИ
- 3. Общая схема кроветворения
- 4. Апластические и гипопластические состояния (приобретенные и врожденные) Апластическая анемия Клональные заболевания пароксизмальная ночная гемоглобинурия миелодиспластический синдром
- 5. Апластические и гипопластические состояния Одноростковая аплазия костного мозга агранулоцитоз, парциальная красноклеточная аплазия, амегокариоцитарная тромбоцитопения
- 6. Young, N. S. Ann Intern Med 2002;136:534-546 Возможная патогенетическая связь синдромов недостаточности костномозгового кроветворения
- 7. Апластическая анемия – заболевание кроветворной ткани, характеризующееся панцитопенией в периферической крови, снижением клеточности костного мозга без
- 8. Copyright ©2008 Ferrata Storti Foundation Montane, E. et al. Haematologica 2008;93:518-523 Table 1. Incidence of aplastic
- 9. Этиологическая классификация апластической анемии Приобретенная Идиопатическая Лекарственно обусловленная Обусловленная действием токсинов и химических веществ Постлучевая Обусловленная
- 10. Апластическая анемия диагностируется при условии снижения клеточности костного мозга на 70 и более % и наличии
- 11. При количестве нейтрофилов в крови меньше чем 0.2 x 109 /л, диагностируется сверхтяжелая апластическая анемия.
- 12. Гистологическая картина костного мозга по данным трепанобиопсии при апластической анемии Апластическая анемия Костный мозг в норме
- 13. жировая ткань в гистологическом препарате костного мозга у больного апластической анемией
- 14. В основе патогенеза идиопатической апластической анемии, как правило, лежат иммунные нарушения. Лимфоциты больных апластической анемией ингибируют
- 15. Увеличение Т-супрессоров у больных апластической анемии по данным проточной цитометрии - предиктор эффективности терапии АТГ и
- 16. Генетическая предрасположенность к развитию апластической анемии Больные со сниженной способностью метаболизировать канцерогены и токсины имеют повышенный
- 17. Наследственные заболевания 2-13% 0% Синдром Kostmann (нейтропения) (аут.-рец.) 2% 0% Анемия Diamond-Blackfan (аут.-рец., аут.-домин.) 5% 20%
- 18. Апластическая анемия может быть ассоциирована с вирусными гепатитами (
- 19. Медицинские препараты, с приемом которых ассоциировано развитие апластической анемии по данным международной группы по апластической анемии
- 20. Апластическая анемияКлинические проявления анемический синдром геморрагический синдром, обусловленный тромбоцитопенией инфекции, обусловленные нейтропенией и иммуносупрессивной терапией (глюкокортикоиды!)
- 21. Лечение тяжелой апластической анемии Первая линия терапии Аллогенная трансплантация стволовых клеток от HLA-совместимого донора родственника Иммуносупрессивная
- 22. Copyright ©2007 Ferrata Storti Foundation Locasciulli, A. et al. Haematologica 2007;92:11-18 Figure 1. Actuarial survival of
- 23. Young, N. S. Ann Intern Med 2002;136:534-546 Патофизиология и лечение апластической анемии Провоцирующий фактор Иммунный ответ
- 24. Эффективность лечения больных апластической анемией в зависимости от возраста Young, N. S. Ann Intern Med 2002;136:534-546
- 25. Young, N. S. Ann Intern Med 2002;136:534-546 Патофизиологические механизмы иммуносупрессивной терапии апластической анемии
- 26. Иммуносупрессивная терапия апластической анемии АЛГ или АТГ 10-40 мг/кг/день 4-5 дня Циклоспорин A (12 мг/кг/день для
- 27. Blood, Vol 85, No 12 (June 15). 1995: pp 3367-3377 Эффективность интенсивной иммуносупрессивной терапии
- 28. Частота рецидивов апластической анемии после проведения иммуносупрессивной терапии .
- 29. Интенсивная иммуносупрессивная терапия Атгам 40 мг/кг день 1-4 дня Преднизолон 1 мг/кг 1-10 дней Циклоспорин А
- 30. Copyright ©2009 Ferrata Storti Foundation Scheinberg, P. et al. Haematologica 2009;94:348-354 Figure 3. Overall survival for
- 31. Динамика гематологических показателей у больных апластической анемией после лечения АТГ и циклоспорином А
- 32. Пример циклоспорин А дозозависимой ремиссии у 21 летней больной апластической анемией. Попытка снижения дозы – рецидив.
- 33. Общая выживаемость больных апластической анемией после неродственной HLA-совместимой трансплантации стволовых клеток
- 34. Экспериментальная терапия терапия Mat к Т-лимфоцитам (at CD3+) терапия Mat к рецептору ИЛ-2 большие дозы циклофосфамида
- 35. Brodsky, R. A. et. al. Ann Intern Med 2001;135:477-483 Проектная выживаемость больных тяжелой апластической анемией после
- 36. Brodsky, R. A. et. al. Ann Intern Med 2001;135:477-483 Гематологический ответ на терапию больных тяжелой апластической
- 37. Socie, G. et al. N Engl J Med 1993;329:1152-1157 Частота опухолевых заболеваний у больных апластической анемией
- 38. Общая выживаемость детей, страдающих тяжелой апластической анемией. (B) частота развития МДС/ОМЛ. (C) Частота МДС/ОМЛ у детей,
- 39. Copyright ©2008 Ferrata Storti Foundation Montane, E. et al. Haematologica 2008;93:518-523 Figure 2. A. Effect of
- 40. АНЕМИЯ ФАНКОНИ – редкое аутосомно-рецессивное заболевание, характеризующееся врожденными аномалиями ( пигментацией кожи, гипоплазией почки или селезенки,
- 41. Внешний вид больного анемией Фанкони
- 42. Гематологические проявления анемии Фанкони развиваются в первые годы жизни. Прогрессирующая мегалобластная анемия иногда в сочетании с
- 43. В настоящее время установлены 8 основных генов, участвующих в развитии анемии Фанкони У части больных выявляются
- 44. 68 (XRCC9) 9p13 редко G 42 11p15 редко F 60 6p21-22 12 E 155,162 3p25.3 редко
- 45. Диагноз анемии Фанкони подтверждается лабораторными тестами. DEB (дайбоксибутановый) тест Клетки больных анемией Фанкони чувствительны к агентам,
- 46. Апластическая анемия развивается у 95% больных анемией Фанкони в возрасте 7-9 лет. Эритропоэз имеет черты стрессового
- 47. Лечение апластической анемии у больных с анемией Фанкони Андрогены (эффект 50%) ГМ-КСФ (эффект 80% в отношении
- 48. Миелодиспластический синдром (МДС) – группа биологически и клинически гетерогенных клональных заболеваний кроветворной ткани, характеризующихся неэффективным гемопоэзом,
- 49. ВОЗ классификация МДС (1) Рефрактерная анемия (РА) анемия > 6 месяцев, наличие дисплазии только в эритроидных
- 50. ВОЗ классификация МДС (2) Рефрактерная анемия с избытком бластов (РАИБ-1) Цитопения и Рефрактерная анемия с избытком
- 51. Медиана возраста больных МДС - 69 лет, Частота встречаемости - 4 случая на 100000 населения. В
- 52. Мегалобластный эритропоэз Многоядерные эритроидные предшественники Увеличение кольцевых сидеробластов Измененные нейтрофилы (псевдопельгеров-ские ) Микро-мегакариоциты Уродливые эритроидные предшественники
- 53. Клиническая картина МДС определяется наличием одно-, двух-, трехростковой цитопениии. Как правило доминирует анемический синдром, реже встречается
- 54. Лечение больных МДС Симптоматическое лечение Химиотерапия Иммуносупрессивная терапия Ростовые факторы Препараты уменьшающие апоптоз Препараты подавляющие ангиогенез
- 55. Пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы-Микели) – приобретенное клональное нарушение гемопоэтических стволовых клеток, характеризующееся нестабильностью клеточных мембран
- 56. В основе патогенеза пароксизмальной ночной гемоглобинурии лежат приобретенные соматические мутации (известны 174 соматические мутации) в гемопоэтических
- 57. Продукт гена PIG-A необходим на ранних этапах синтеза гликозилфосфатидилинозитольных структур мембран, которые служат «якорем» для групп
- 58. Проточная цитометрия – основной метод диагностики ПНГ, так как позволяет оценить экспрессию GPI-якорных белков. Вспомогательное значение
- 59. Dunn, D. E. et. al. Ann Intern Med 1999;131:401-408 Выраженность гемолиза эритроцитов при пробе Хема в
- 60. CD55 и CD59 могут использоваться для анализа экспрессии GPI-якорных протеинов на эритроцитах. Гликофорин А – неякорный
- 61. Dunn, D. E. et. al. Ann Intern Med 1999;131:401-408 Протокол идентификации пароксизмальной ночной гемоглобинурии с помощью
- 62. Клинические симптомы у больных с пароксизмальной ночной гемоглобинурией зависят от степени вовлечения мегакариоцитарного и гранулоцитарного ростков
- 63. Клиническая гетерогенность ПНГ Мини-ПНГ маленький Отсутствие или умеренная цитопения тромбоз Апластическая анемия с ПНГ клоном маленький
- 64. Рекомендации для лечения ПНГ Диагноз ПНГ определение клинической формы Флоридская ПНГ гипопластическая АА,ПНГ АА с ПНГ
- 65. Парциальная красноклеточная аплазия/ чистая красноклеточная аплазия (ЧККА) – клинический синдром, характеризующийся отсутствием зрелых эритроидных предшественников на
- 66. Патогенез парциальной красноклеточной аплазии T-лимфоцит опосредованная супрессия эритропоэза антителообусловленная супрессия эритропоэза смешанный механизм
- 67. Патогенез парциальной красноклеточной аплазии
- 68. Блок созревания эритроидных предшественников 100% 63% 22% 0% Blood, Vol 87, No 11 (June 1). 1996:
- 69. Этиологическая классификация парциальной красноклеточной аплазии Врожденная гипопластическая анемия (синдром Даймонда-Блекфана) Приобретенная А. Первичная 1. Аутоиммунная 2.
- 70. Б. Вторичная ЧККА, связанная с различными заболеваниями и состояниями: Тимома Онкогематологические заболевания (Т-, В-ХЛЛ, лимфома/лейкоз из
- 71. Иммуносупрессивная терапия парциальной красноклеточной аплазии Первая линия Преднизолон Вторая линия Циклофосфамид Циклоспорин
- 72. Ответ на лечение преднизолоном и циклофосфамидом больного парциальной красноклеточной аплазией
- 73. Агранулоцитоз – наличие у больного тяжелой (IV степени) нейтропении (количество нейтрофилов в крови
- 74. Этиологическая классификация нейтропений (приобретенные и врожденные) I. Приобретенные нейтропении 1)IИнфекции (наиболее частая причина) Вирусные Бактериальные Протозойные
- 75. I. Приобретенные нейтропении 2) Лекарство- или химически опосредованные: - соли тяжелых металлов - анальгетики, нестероидные противовоспалительные
- 76. Приобретенные нейтропении 3) Иммунные нейтропении Изоиммунная неонатальная нейтропения Хроническая аутоиммунная нейтропения Tγ – лимфоцитоз Смешанные иммуноопосредованные
- 77. II. Врожденные нейтропении 1) Тяжелая врожденная нейтропения (синдром Костмана) 2) Циклическая нейтропения 3) Хроническая доброкачественная нейтропения
- 79. Скачать презентацию

ПРИЧИНЫ ЦИТОПЕНИЧЕСКИХ СОСТОЯНИЙ
НАРУШЕНИЕ ПРОДУКЦИИ КЛЕТОК КРОВИ
ПОВЫШЕННОЕ РАЗРУШЕНИЕ КЛЕТОК КРОВИ
ПЕРЕРАСПРЕДЕЛЕНИЕ КЛЕТОК КРОВИ
ПРИЧИНЫ ЦИТОПЕНИЧЕСКИХ СОСТОЯНИЙ
НАРУШЕНИЕ ПРОДУКЦИИ КЛЕТОК КРОВИ
ПОВЫШЕННОЕ РАЗРУШЕНИЕ КЛЕТОК КРОВИ
ПЕРЕРАСПРЕДЕЛЕНИЕ КЛЕТОК КРОВИ
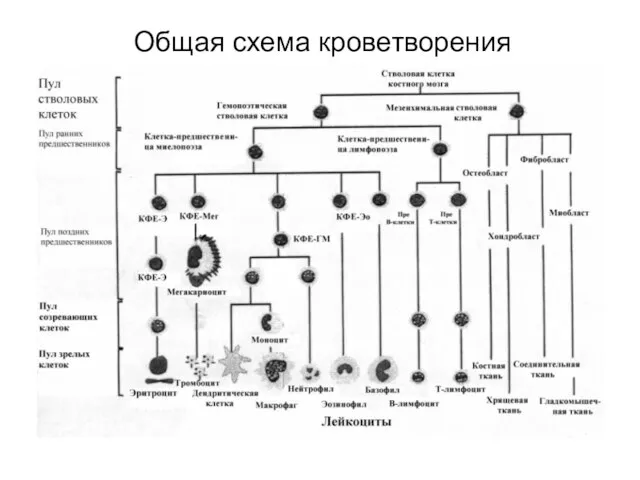
Общая схема кроветворения
Общая схема кроветворения

Апластические и гипопластические состояния (приобретенные и врожденные)
Апластическая анемия
Клональные заболевания
пароксизмальная ночная гемоглобинурия
миелодиспластический
Апластические и гипопластические состояния (приобретенные и врожденные)
Апластическая анемия
Клональные заболевания
пароксизмальная ночная гемоглобинурия
миелодиспластический

Апластические и гипопластические состояния
Одноростковая аплазия костного мозга
агранулоцитоз,
парциальная красноклеточная аплазия,
Апластические и гипопластические состояния
Одноростковая аплазия костного мозга
агранулоцитоз,
парциальная красноклеточная аплазия,
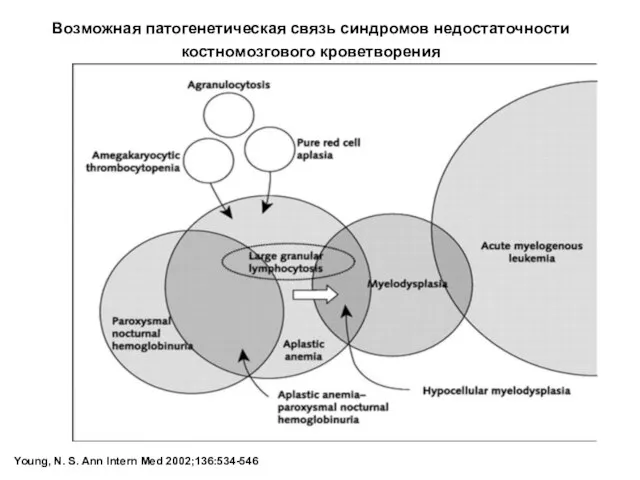
Young, N. S. Ann Intern Med 2002;136:534-546
Возможная патогенетическая связь синдромов недостаточности
Young, N. S. Ann Intern Med 2002;136:534-546
Возможная патогенетическая связь синдромов недостаточности

Апластическая анемия – заболевание кроветворной ткани, характеризующееся панцитопенией в периферической крови,
Апластическая анемия – заболевание кроветворной ткани, характеризующееся панцитопенией в периферической крови,
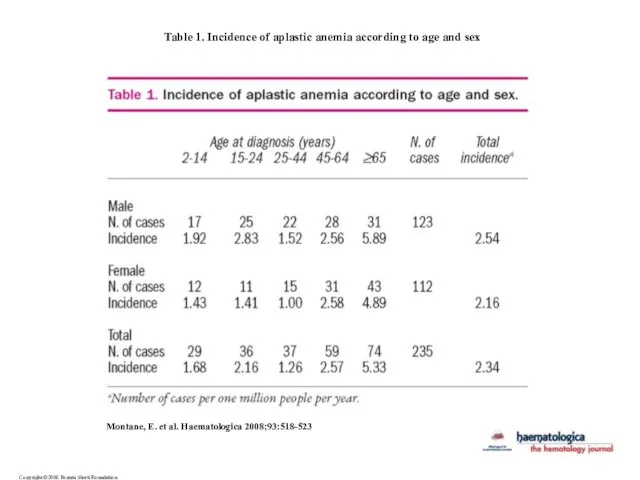
Copyright ©2008 Ferrata Storti Foundation
Montane, E. et al. Haematologica 2008;93:518-523
Table 1.
Copyright ©2008 Ferrata Storti Foundation
Montane, E. et al. Haematologica 2008;93:518-523
Table 1.

Этиологическая классификация апластической анемии
Приобретенная
Идиопатическая
Лекарственно обусловленная
Обусловленная действием токсинов и химических веществ
Постлучевая
Обусловленная инфекцией
Гепатит
Парвовирус
ВИЧ
Беременность
Тимома
Ассоциированная
Этиологическая классификация апластической анемии
Приобретенная
Идиопатическая
Лекарственно обусловленная
Обусловленная действием токсинов и химических веществ
Постлучевая
Обусловленная инфекцией
Гепатит
Парвовирус
ВИЧ
Беременность
Тимома
Ассоциированная

Апластическая анемия диагностируется при условии снижения клеточности костного мозга на 70
Апластическая анемия диагностируется при условии снижения клеточности костного мозга на 70

При количестве нейтрофилов в крови меньше чем 0.2 x 109 /л,
При количестве нейтрофилов в крови меньше чем 0.2 x 109 /л,
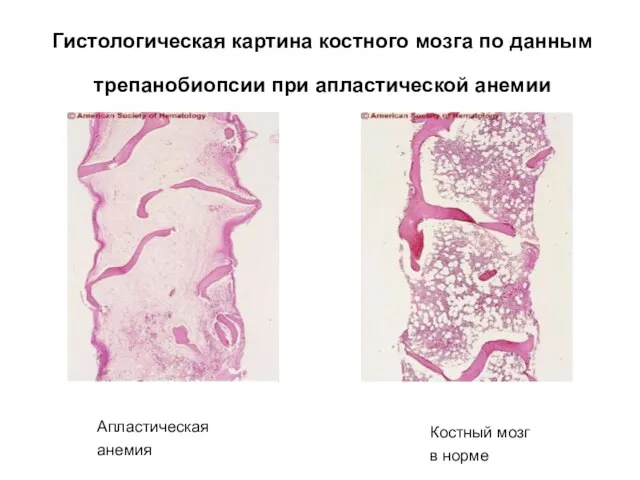
Гистологическая картина костного мозга по данным трепанобиопсии при апластической анемии
Апластическая
Гистологическая картина костного мозга по данным трепанобиопсии при апластической анемии
Апластическая
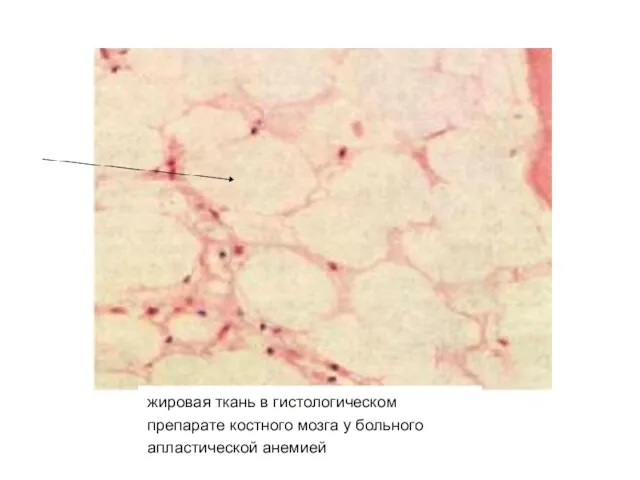
жировая ткань в гистологическом препарате костного мозга у больного апластической анемией
жировая ткань в гистологическом препарате костного мозга у больного апластической анемией

В основе патогенеза идиопатической апластической анемии, как правило, лежат иммунные нарушения.
В основе патогенеза идиопатической апластической анемии, как правило, лежат иммунные нарушения.
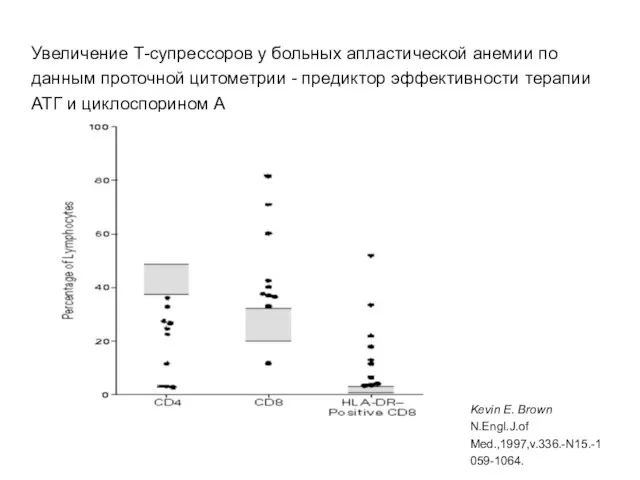
Увеличение Т-супрессоров у больных апластической анемии по данным проточной цитометрии -
Увеличение Т-супрессоров у больных апластической анемии по данным проточной цитометрии -

Генетическая предрасположенность к развитию апластической анемии
Больные со сниженной способностью метаболизировать канцерогены
Генетическая предрасположенность к развитию апластической анемии
Больные со сниженной способностью метаболизировать канцерогены
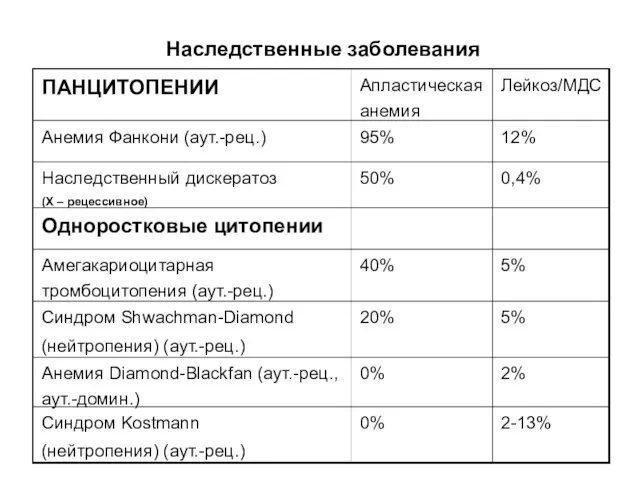
Наследственные заболевания
2-13%
0%
Синдром Kostmann
(нейтропения) (аут.-рец.)
2%
0%
Анемия Diamond-Blackfan (аут.-рец., аут.-домин.)
5%
20%
Синдром Shwachman-Diamond
(нейтропения) (аут.-рец.)
5%
40%
Амегакариоцитарная тромбоцитопения
Наследственные заболевания
2-13%
0%
Синдром Kostmann
(нейтропения) (аут.-рец.)
2%
0%
Анемия Diamond-Blackfan (аут.-рец., аут.-домин.)
5%
20%
Синдром Shwachman-Diamond
(нейтропения) (аут.-рец.)
5%
40%
Амегакариоцитарная тромбоцитопения

Апластическая анемия может быть ассоциирована с вирусными гепатитами (< 1% случаев)
Апластическая анемия может быть ассоциирована с вирусными гепатитами (< 1% случаев)

Медицинские препараты, с приемом которых ассоциировано развитие апластической анемии по данным
Медицинские препараты, с приемом которых ассоциировано развитие апластической анемии по данным

Апластическая анемияКлинические проявления
анемический синдром
геморрагический синдром, обусловленный тромбоцитопенией
инфекции, обусловленные нейтропенией и
Апластическая анемияКлинические проявления
анемический синдром
геморрагический синдром, обусловленный тромбоцитопенией
инфекции, обусловленные нейтропенией и

Лечение тяжелой апластической анемии
Первая линия терапии
Аллогенная трансплантация стволовых клеток от HLA-совместимого
Лечение тяжелой апластической анемии
Первая линия терапии
Аллогенная трансплантация стволовых клеток от HLA-совместимого
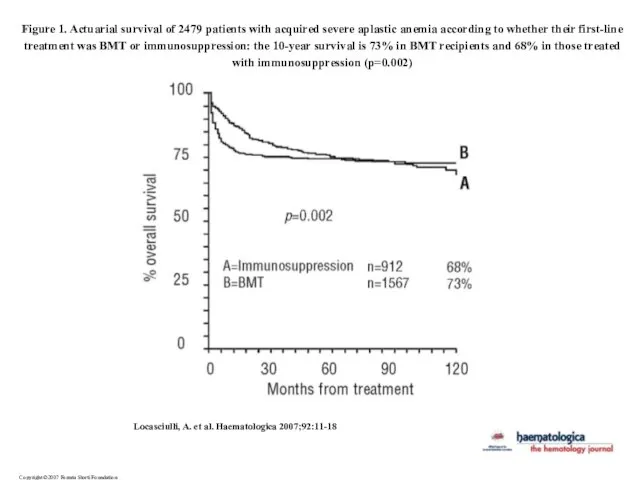
Copyright ©2007 Ferrata Storti Foundation
Locasciulli, A. et al. Haematologica 2007;92:11-18
Figure 1.
Copyright ©2007 Ferrata Storti Foundation
Locasciulli, A. et al. Haematologica 2007;92:11-18
Figure 1.
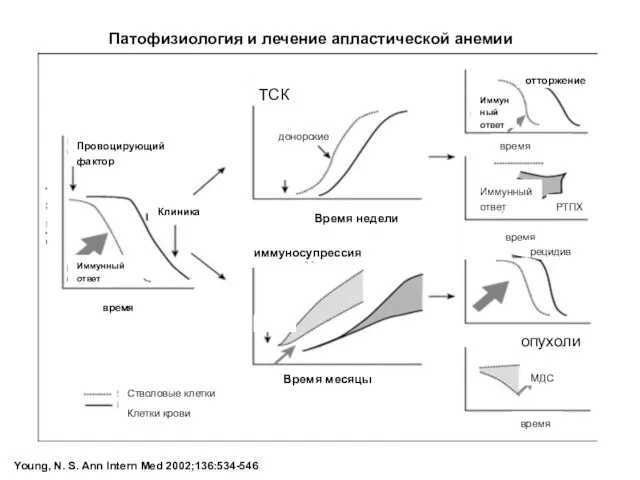
Young, N. S. Ann Intern Med 2002;136:534-546
Патофизиология и лечение апластической анемии
Провоцирующий
Young, N. S. Ann Intern Med 2002;136:534-546
Патофизиология и лечение апластической анемии
Провоцирующий
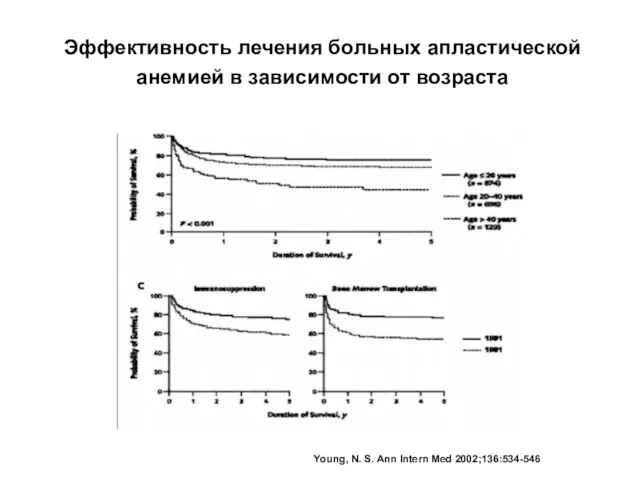
Эффективность лечения больных апластической анемией в зависимости от возраста
Young, N. S.
Эффективность лечения больных апластической анемией в зависимости от возраста
Young, N. S.
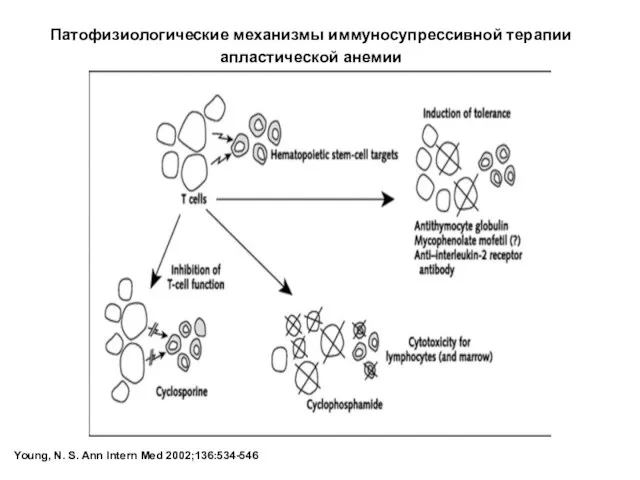
Young, N. S. Ann Intern Med 2002;136:534-546
Патофизиологические механизмы иммуносупрессивной терапии апластической
Young, N. S. Ann Intern Med 2002;136:534-546
Патофизиологические механизмы иммуносупрессивной терапии апластической

Иммуносупрессивная терапия апластической анемии
АЛГ или АТГ 10-40 мг/кг/день 4-5 дня
Циклоспорин A
Иммуносупрессивная терапия апластической анемии
АЛГ или АТГ 10-40 мг/кг/день 4-5 дня
Циклоспорин A
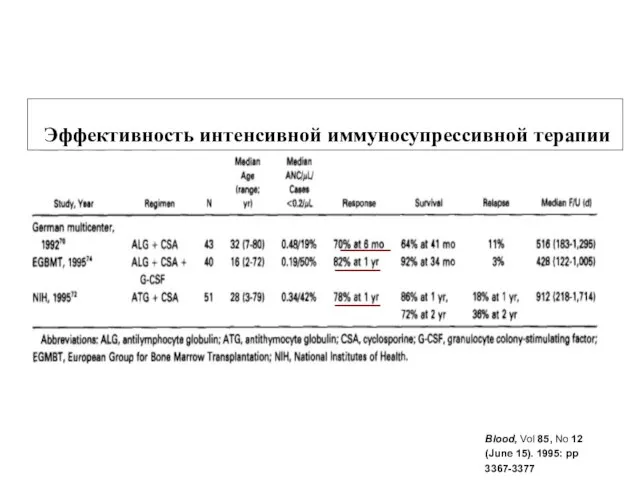
Blood, Vol 85, No 12 (June 15). 1995: pp 3367-3377
Эффективность интенсивной
Blood, Vol 85, No 12 (June 15). 1995: pp 3367-3377
Эффективность интенсивной
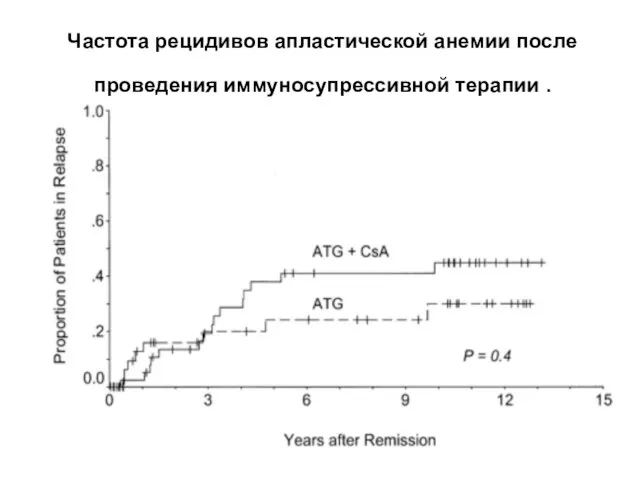
Частота рецидивов апластической анемии после проведения иммуносупрессивной терапии .
Частота рецидивов апластической анемии после проведения иммуносупрессивной терапии .

Интенсивная иммуносупрессивная терапия
Атгам 40 мг/кг день 1-4 дня
Преднизолон 1 мг/кг 1-10
Интенсивная иммуносупрессивная терапия
Атгам 40 мг/кг день 1-4 дня
Преднизолон 1 мг/кг 1-10
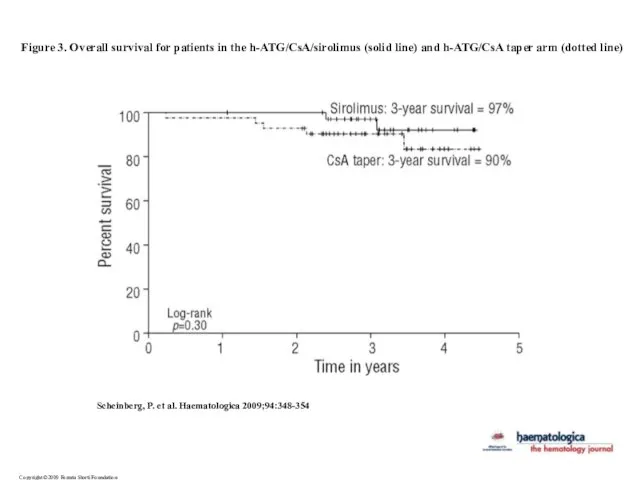
Copyright ©2009 Ferrata Storti Foundation
Scheinberg, P. et al. Haematologica 2009;94:348-354
Figure 3.
Copyright ©2009 Ferrata Storti Foundation
Scheinberg, P. et al. Haematologica 2009;94:348-354
Figure 3.
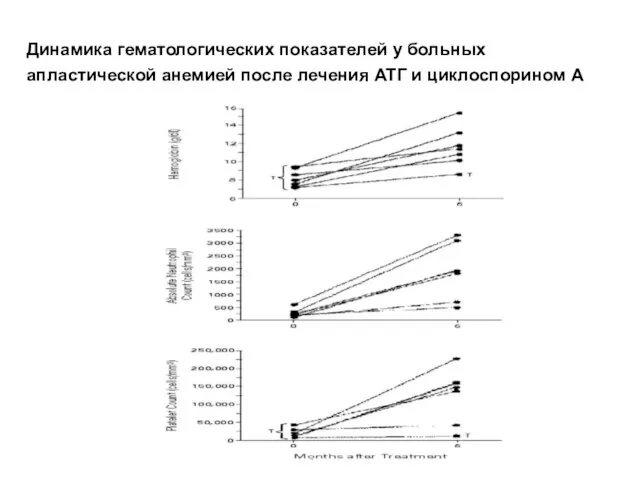
Динамика гематологических показателей у больных апластической анемией после лечения АТГ и
Динамика гематологических показателей у больных апластической анемией после лечения АТГ и
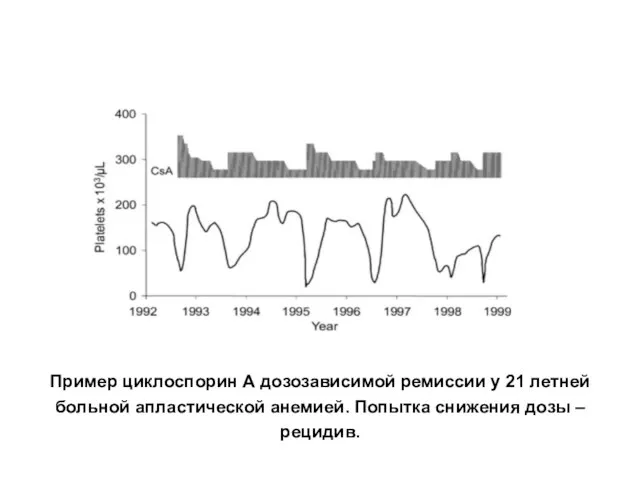
Пример циклоспорин А дозозависимой ремиссии у 21 летней больной апластической анемией.
Пример циклоспорин А дозозависимой ремиссии у 21 летней больной апластической анемией.
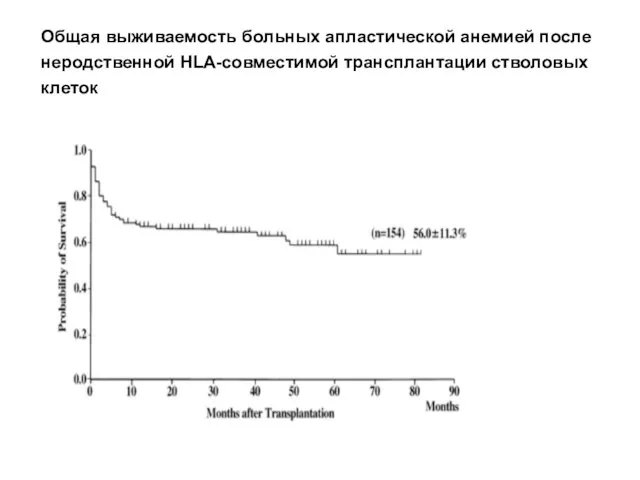
Общая выживаемость больных апластической анемией после неродственной HLA-совместимой трансплантации стволовых клеток
Общая выживаемость больных апластической анемией после неродственной HLA-совместимой трансплантации стволовых клеток

Экспериментальная терапия
терапия Mat к Т-лимфоцитам (at CD3+)
терапия Mat к рецептору ИЛ-2
большие
Экспериментальная терапия
терапия Mat к Т-лимфоцитам (at CD3+)
терапия Mat к рецептору ИЛ-2
большие
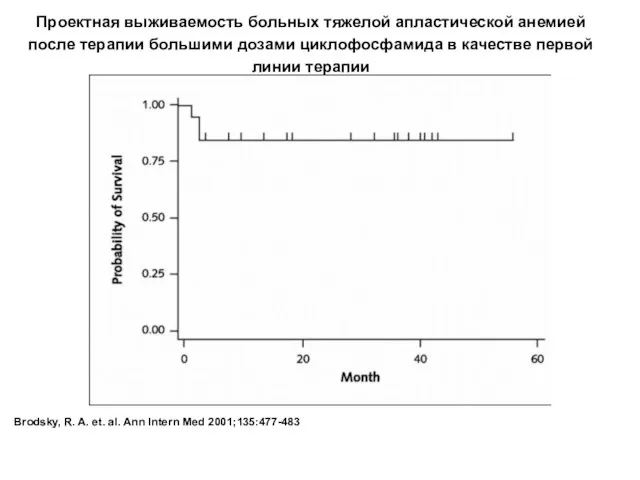
Brodsky, R. A. et. al. Ann Intern Med 2001;135:477-483
Проектная выживаемость больных
Brodsky, R. A. et. al. Ann Intern Med 2001;135:477-483
Проектная выживаемость больных

Brodsky, R. A. et. al. Ann Intern Med 2001;135:477-483
Гематологический ответ на
Brodsky, R. A. et. al. Ann Intern Med 2001;135:477-483
Гематологический ответ на
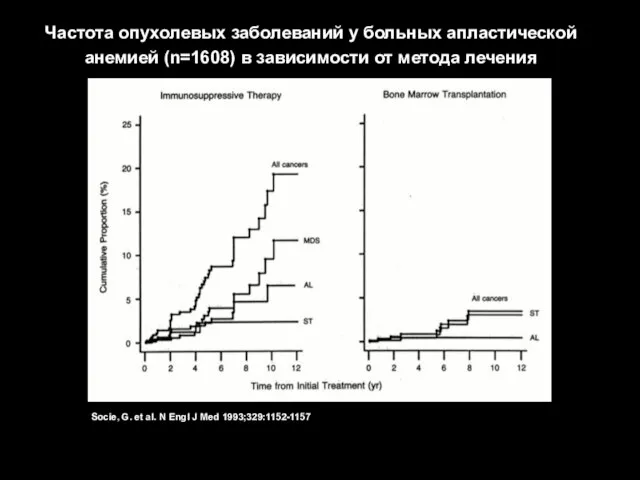
Socie, G. et al. N Engl J Med 1993;329:1152-1157
Частота опухолевых заболеваний
Socie, G. et al. N Engl J Med 1993;329:1152-1157
Частота опухолевых заболеваний
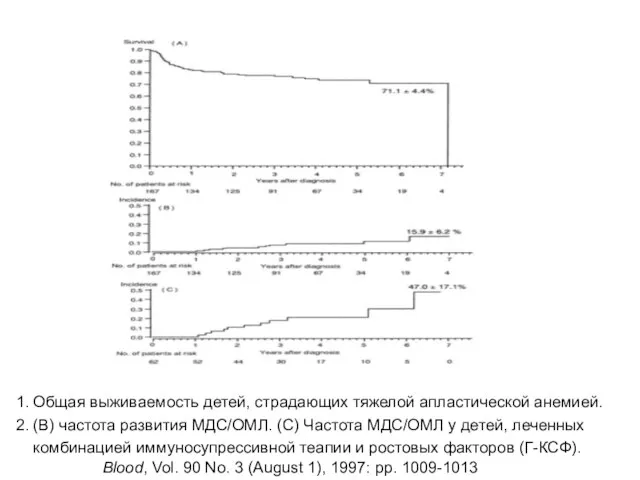
Общая выживаемость детей, страдающих тяжелой апластической анемией.
(B) частота развития МДС/ОМЛ.
Общая выживаемость детей, страдающих тяжелой апластической анемией.
(B) частота развития МДС/ОМЛ.
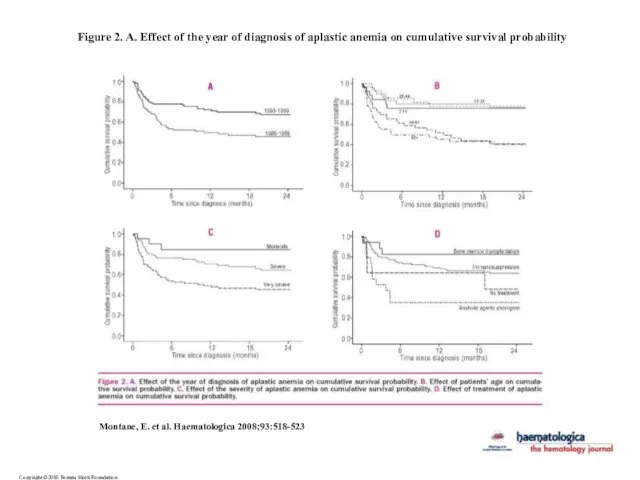
Copyright ©2008 Ferrata Storti Foundation
Montane, E. et al. Haematologica 2008;93:518-523
Figure 2.
Copyright ©2008 Ferrata Storti Foundation
Montane, E. et al. Haematologica 2008;93:518-523
Figure 2.

АНЕМИЯ ФАНКОНИ – редкое аутосомно-рецессивное заболевание, характеризующееся врожденными аномалиями ( пигментацией
АНЕМИЯ ФАНКОНИ – редкое аутосомно-рецессивное заболевание, характеризующееся врожденными аномалиями ( пигментацией

Внешний вид больного анемией Фанкони
Внешний вид больного анемией Фанкони

Гематологические проявления анемии Фанкони развиваются в первые годы жизни. Прогрессирующая мегалобластная
Гематологические проявления анемии Фанкони развиваются в первые годы жизни. Прогрессирующая мегалобластная

В настоящее время установлены 8 основных генов, участвующих в развитии анемии
В настоящее время установлены 8 основных генов, участвующих в развитии анемии
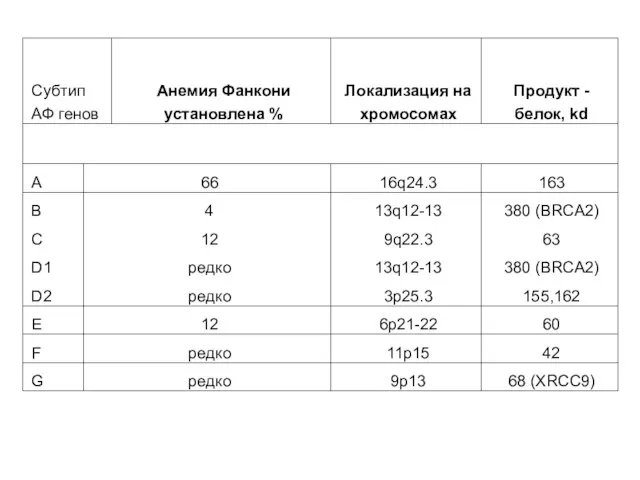
68 (XRCC9)
9p13
редко
G
42
11p15
редко
F
60
6p21-22
12
E
155,162
3p25.3
редко
D2
380 (BRCA2)
13q12-13
редко
D1
63
9q22.3
12
C
380 (BRCA2)
13q12-13
4
B
163
16q24.3
66
A
Продукт - белок, kd
Локализация на хромосомах
Анемия Фанкони установлена
68 (XRCC9)
9p13
редко
G
42
11p15
редко
F
60
6p21-22
12
E
155,162
3p25.3
редко
D2
380 (BRCA2)
13q12-13
редко
D1
63
9q22.3
12
C
380 (BRCA2)
13q12-13
4
B
163
16q24.3
66
A
Продукт - белок, kd
Локализация на хромосомах
Анемия Фанкони установлена

Диагноз анемии Фанкони подтверждается лабораторными тестами.
DEB (дайбоксибутановый) тест
Клетки больных анемией Фанкони
Диагноз анемии Фанкони подтверждается лабораторными тестами.
DEB (дайбоксибутановый) тест
Клетки больных анемией Фанкони

Апластическая анемия развивается у 95% больных анемией Фанкони в возрасте 7-9
Апластическая анемия развивается у 95% больных анемией Фанкони в возрасте 7-9

Лечение апластической анемии у больных с анемией Фанкони
Андрогены (эффект 50%)
ГМ-КСФ
Лечение апластической анемии у больных с анемией Фанкони
Андрогены (эффект 50%)
ГМ-КСФ

Миелодиспластический синдром (МДС) – группа биологически и клинически гетерогенных клональных заболеваний
Миелодиспластический синдром (МДС) – группа биологически и клинически гетерогенных клональных заболеваний

ВОЗ классификация МДС (1)
Рефрактерная анемия (РА)
анемия > 6 месяцев, наличие
ВОЗ классификация МДС (1)
Рефрактерная анемия (РА)
анемия > 6 месяцев, наличие

ВОЗ классификация МДС (2)
Рефрактерная анемия с избытком бластов (РАИБ-1)
Цитопения и <
ВОЗ классификация МДС (2)
Рефрактерная анемия с избытком бластов (РАИБ-1)
Цитопения и <

Медиана возраста больных МДС - 69 лет,
Частота встречаемости - 4 случая
Медиана возраста больных МДС - 69 лет,
Частота встречаемости - 4 случая
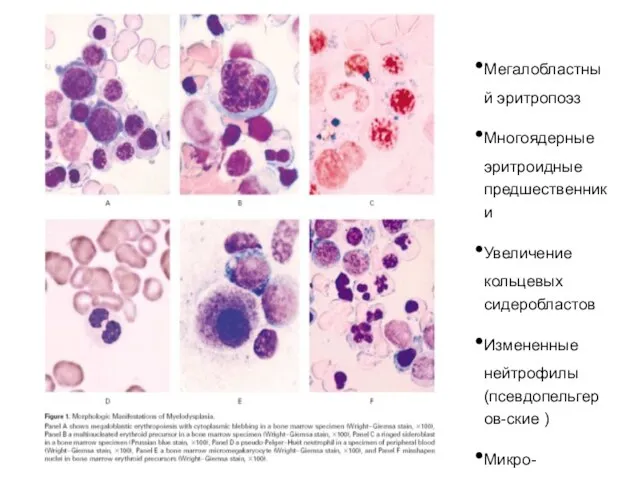
Мегалобластный эритропоэз
Многоядерные эритроидные предшественники
Увеличение кольцевых сидеробластов
Измененные нейтрофилы (псевдопельгеров-ские )
Микро-мегакариоциты
Уродливые эритроидные предшественники
Мегалобластный эритропоэз
Многоядерные эритроидные предшественники
Увеличение кольцевых сидеробластов
Измененные нейтрофилы (псевдопельгеров-ские )
Микро-мегакариоциты
Уродливые эритроидные предшественники

Клиническая картина МДС определяется наличием одно-, двух-, трехростковой цитопениии. Как правило
Клиническая картина МДС определяется наличием одно-, двух-, трехростковой цитопениии. Как правило

Лечение больных МДС
Симптоматическое лечение
Химиотерапия
Иммуносупрессивная терапия
Ростовые факторы
Препараты уменьшающие апоптоз
Препараты подавляющие ангиогенез
Трансплантация стволовых
Лечение больных МДС
Симптоматическое лечение
Химиотерапия
Иммуносупрессивная терапия
Ростовые факторы
Препараты уменьшающие апоптоз
Препараты подавляющие ангиогенез
Трансплантация стволовых

Пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы-Микели) –
приобретенное клональное нарушение гемопоэтических стволовых
Пароксизмальная ночная гемоглобинурия (болезнь Маркиафавы-Микели) –
приобретенное клональное нарушение гемопоэтических стволовых

В основе патогенеза пароксизмальной ночной гемоглобинурии лежат приобретенные соматические мутации (известны
В основе патогенеза пароксизмальной ночной гемоглобинурии лежат приобретенные соматические мутации (известны

Продукт гена PIG-A необходим на ранних этапах синтеза гликозилфосфатидилинозитольных структур мембран,
Продукт гена PIG-A необходим на ранних этапах синтеза гликозилфосфатидилинозитольных структур мембран,

Проточная цитометрия – основной метод диагностики ПНГ, так как позволяет оценить
Проточная цитометрия – основной метод диагностики ПНГ, так как позволяет оценить
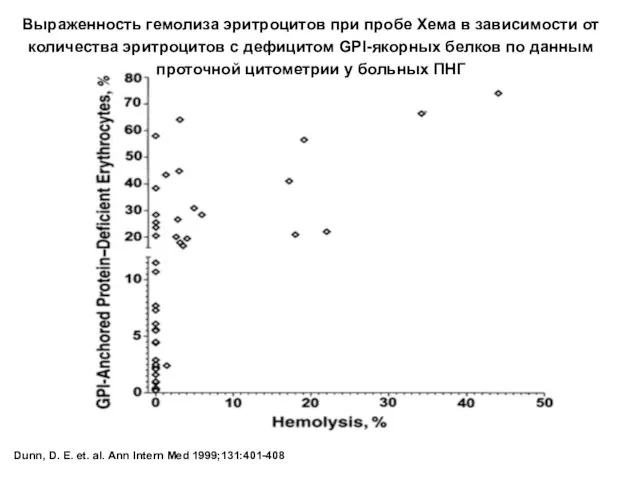
Dunn, D. E. et. al. Ann Intern Med 1999;131:401-408
Выраженность гемолиза эритроцитов
Dunn, D. E. et. al. Ann Intern Med 1999;131:401-408
Выраженность гемолиза эритроцитов

CD55 и CD59 могут использоваться для анализа экспрессии GPI-якорных протеинов на
CD55 и CD59 могут использоваться для анализа экспрессии GPI-якорных протеинов на
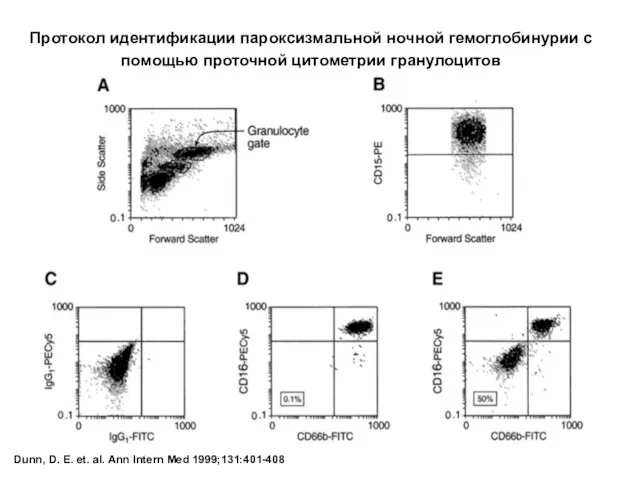
Dunn, D. E. et. al. Ann Intern Med 1999;131:401-408
Протокол идентификации пароксизмальной
Dunn, D. E. et. al. Ann Intern Med 1999;131:401-408
Протокол идентификации пароксизмальной

Клинические симптомы у больных с пароксизмальной ночной гемоглобинурией зависят от степени
Клинические симптомы у больных с пароксизмальной ночной гемоглобинурией зависят от степени
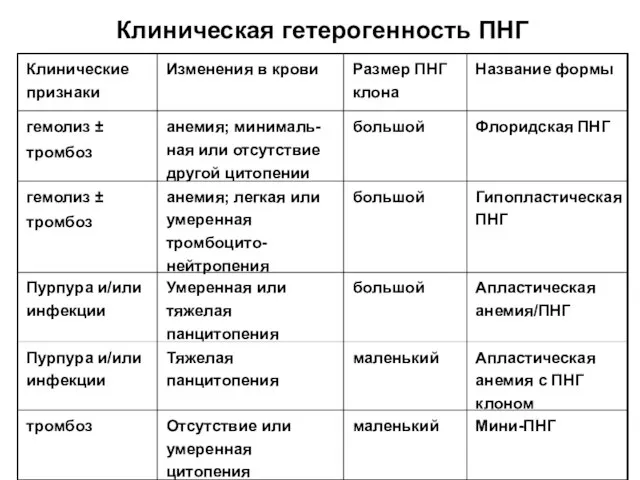
Клиническая гетерогенность ПНГ
Мини-ПНГ
маленький
Отсутствие или умеренная цитопения
тромбоз
Апластическая анемия с ПНГ клоном
маленький
Тяжелая панцитопения
Пурпура
Клиническая гетерогенность ПНГ
Мини-ПНГ
маленький
Отсутствие или умеренная цитопения
тромбоз
Апластическая анемия с ПНГ клоном
маленький
Тяжелая панцитопения
Пурпура
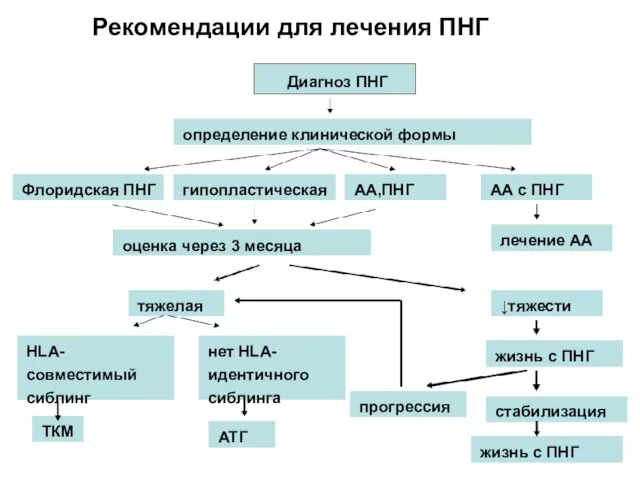
Рекомендации для лечения ПНГ
Диагноз ПНГ
определение клинической формы
Флоридская ПНГ
гипопластическая
АА,ПНГ
АА с ПНГ
оценка
Рекомендации для лечения ПНГ
Диагноз ПНГ
определение клинической формы
Флоридская ПНГ
гипопластическая
АА,ПНГ
АА с ПНГ
оценка

Парциальная красноклеточная аплазия/
чистая красноклеточная аплазия (ЧККА) –
клинический синдром, характеризующийся
отсутствием зрелых эритроидных
предшественников
Парциальная красноклеточная аплазия/
чистая красноклеточная аплазия (ЧККА) –
клинический синдром, характеризующийся
отсутствием зрелых эритроидных
предшественников

Патогенез парциальной красноклеточной аплазии
T-лимфоцит опосредованная супрессия эритропоэза
антителообусловленная супрессия эритропоэза
смешанный механизм
Патогенез парциальной красноклеточной аплазии
T-лимфоцит опосредованная супрессия эритропоэза
антителообусловленная супрессия эритропоэза
смешанный механизм
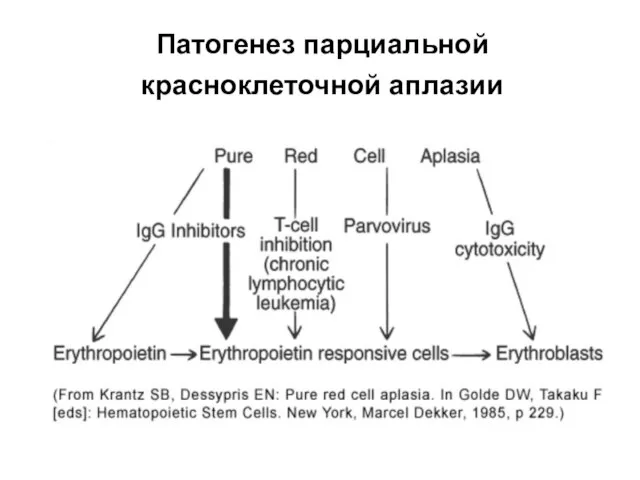
Патогенез парциальной красноклеточной аплазии
Патогенез парциальной красноклеточной аплазии
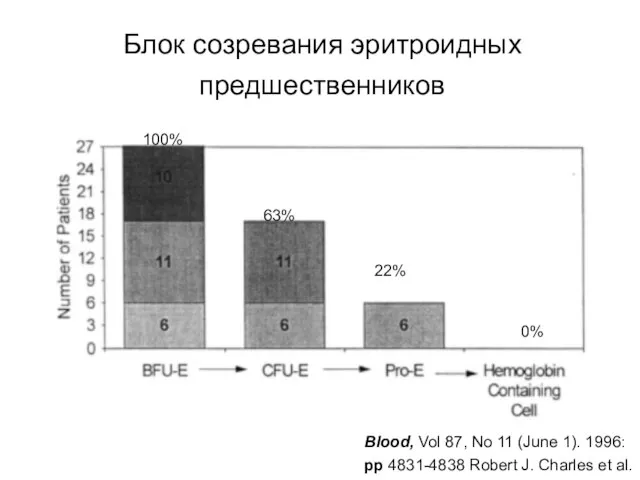
Блок созревания эритроидных предшественников
100%
63%
22%
0%
Blood, Vol 87, No 11 (June 1). 1996:
Блок созревания эритроидных предшественников
100%
63%
22%
0%
Blood, Vol 87, No 11 (June 1). 1996:

Этиологическая классификация парциальной красноклеточной аплазии
Врожденная гипопластическая анемия (синдром Даймонда-Блекфана)
Приобретенная
А. Первичная
1. Аутоиммунная
2.
Этиологическая классификация парциальной красноклеточной аплазии
Врожденная гипопластическая анемия (синдром Даймонда-Блекфана)
Приобретенная
А. Первичная
1. Аутоиммунная
2.

Б. Вторичная ЧККА, связанная с различными заболеваниями и состояниями:
Тимома
Онкогематологические заболевания (Т-,
Б. Вторичная ЧККА, связанная с различными заболеваниями и состояниями:
Тимома
Онкогематологические заболевания (Т-,

Иммуносупрессивная терапия парциальной красноклеточной аплазии
Первая линия
Преднизолон
Вторая линия
Циклофосфамид
Циклоспорин
Иммуносупрессивная терапия парциальной красноклеточной аплазии
Первая линия
Преднизолон
Вторая линия
Циклофосфамид
Циклоспорин
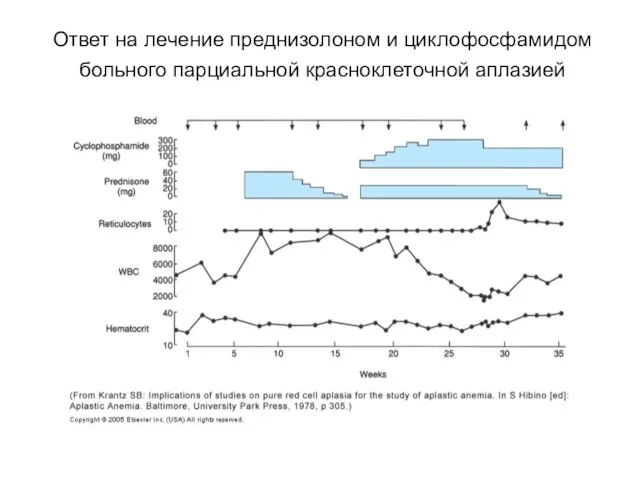
Ответ на лечение преднизолоном и циклофосфамидом больного парциальной красноклеточной аплазией
Ответ на лечение преднизолоном и циклофосфамидом больного парциальной красноклеточной аплазией

Агранулоцитоз – наличие у больного тяжелой (IV степени) нейтропении (количество нейтрофилов
Агранулоцитоз – наличие у больного тяжелой (IV степени) нейтропении (количество нейтрофилов

Этиологическая классификация нейтропений
(приобретенные и врожденные)
I. Приобретенные нейтропении
1)IИнфекции (наиболее частая причина)
Вирусные
Бактериальные
Протозойные
Риккетсиозные
Грибковые
(нейтропения
Этиологическая классификация нейтропений
(приобретенные и врожденные)
I. Приобретенные нейтропении
1)IИнфекции (наиболее частая причина)
Вирусные
Бактериальные
Протозойные
Риккетсиозные
Грибковые
(нейтропения

I. Приобретенные нейтропении
2) Лекарство- или химически опосредованные:
- соли тяжелых металлов
-
I. Приобретенные нейтропении
2) Лекарство- или химически опосредованные:
- соли тяжелых металлов
-

Приобретенные нейтропении
3) Иммунные нейтропении
Изоиммунная неонатальная нейтропения
Хроническая аутоиммунная нейтропения
Tγ – лимфоцитоз
Смешанные иммуноопосредованные
Приобретенные нейтропении
3) Иммунные нейтропении
Изоиммунная неонатальная нейтропения
Хроническая аутоиммунная нейтропения
Tγ – лимфоцитоз
Смешанные иммуноопосредованные

II. Врожденные нейтропении
1) Тяжелая врожденная нейтропения (синдром Костмана)
2) Циклическая нейтропения
3) Хроническая
II. Врожденные нейтропении
1) Тяжелая врожденная нейтропения (синдром Костмана)
2) Циклическая нейтропения
3) Хроническая
 Судороги у детей, дифференциальная диагностика
Судороги у детей, дифференциальная диагностика Лимфодренажная гимнастика. Лимфодренажный самомассаж. Массаж ложками
Лимфодренажная гимнастика. Лимфодренажный самомассаж. Массаж ложками Обезболивание в клинике ортопедической стоматологии. Методы обезболивания
Обезболивание в клинике ортопедической стоматологии. Методы обезболивания Невербальная коммуникация
Невербальная коммуникация Консервативное лечение пролапса тазовых органов. Краткий обзор методов нехирургического дизайна промежности
Консервативное лечение пролапса тазовых органов. Краткий обзор методов нехирургического дизайна промежности Психологические особенности детей с нарушениями речи
Психологические особенности детей с нарушениями речи Сестринский уход при заболеваниях придаточного аппарата глаза и оболочек глазного яблока
Сестринский уход при заболеваниях придаточного аппарата глаза и оболочек глазного яблока Аденовирусная инфекция крупного рогатого скота (аденовирусная пневмония телят, аденовирусный пневмоэнтерит телят)
Аденовирусная инфекция крупного рогатого скота (аденовирусная пневмония телят, аденовирусный пневмоэнтерит телят) Кетоацидоздық кома
Кетоацидоздық кома Васкулиты кожи
Васкулиты кожи Моя профессия ветеринар
Моя профессия ветеринар Анатомия живого человека
Анатомия живого человека методы обследования больных. Анамнестическая часть истории болезни. Осмотр больного и его значение в диагностическом процессе
методы обследования больных. Анамнестическая часть истории болезни. Осмотр больного и его значение в диагностическом процессе Корректные методы убеждений партнёра
Корректные методы убеждений партнёра Лечение острого коронарного синдрома. Консервативная стратегия
Лечение острого коронарного синдрома. Консервативная стратегия Биологические свойства возбудителей дизентерии
Биологические свойства возбудителей дизентерии Абдоминальный ишемический синдром
Абдоминальный ишемический синдром Вирусные диареи
Вирусные диареи Школа здоровья для беременных. Занятие 3. Как себя вести: вопросы и ответы
Школа здоровья для беременных. Занятие 3. Как себя вести: вопросы и ответы Прививая литературу. Русские писатели-врачи
Прививая литературу. Русские писатели-врачи Что такое ТМС?
Что такое ТМС? Сон и сновидения
Сон и сновидения Способы удовлетворения основных потребностей гериатрического пациента
Способы удовлетворения основных потребностей гериатрического пациента Введение в психоанализ
Введение в психоанализ Бехтерев ауруы
Бехтерев ауруы Инфекциялық әлеуметтік маңызы бар аурулар. Аса қауіпті инфекциялар (жұқпалар). Туберкулез
Инфекциялық әлеуметтік маңызы бар аурулар. Аса қауіпті инфекциялар (жұқпалар). Туберкулез Болезни сердца
Болезни сердца Організація медико-соціального забезпечення населення літнього віку
Організація медико-соціального забезпечення населення літнього віку