- Изменчивость. Формы изменчивости организмов

Содержание
- 4. Изменчивость свойство живых организмов приобретать в процессе индивидуального развития новые признаки и свойства
- 5. Формы изменчивости организмов
- 6. Формы изменчивости Наследственная мутационная комбинационная цитоплазматическая Ненаследственная модификационная фенотипическая
- 7. ген белок признак генотип фенотип Факторы окружающей среды
- 8. Изменчивость организма, возникающая под влиянием факторов внешней среды и не затрагивающая генотип, называется модификационной Модификация –
- 9. Вариационные кривые изменчивости морфофункциональных параметров школьников
- 10. Норма реакции – степень варьирования признака или пределы модификационной изменчивости, обусловленные генотипом Наследуется не признак как
- 11. Выводы: 1. Проявление признака не выходит за пределы нормы реакции, которая определяется генотипом. 2. Среди показателей
- 12. Основные характеристики модификационной изменчивости 1. Зависит от окружающих условий. 2. Носит групповой характер. 3. Является определённой.
- 13. Средняя величина признака V1 х p1 + V2 х p2+ Vn х pn М= m М
- 14. Наследственная (генотипическая) изменчивость Комбинативная Мутационная Генная Хромосомная Геномная
- 15. Морфоз (случайная фенотипическая изменчивость) – необратимые изменения фенотипа, которые человек получает в течение жизни. Возникает под
- 16. Характкристики: 1. Не имеет приспособительного характера; 2. Возникает под действием неблагоприятных или экстремальных факторов внешней среды;
- 17. Пенетрантность – процент реализации гена в признак. Пенетрантность выражается отношением числа особей у которых проявляется признак,
- 18. Фенокопия — ненаследуемое изменение фенотипа, которое возникает под влиянием внешних факторов и своим проявлением подобно мутации,
- 19. Сезонная изменчивость — это внешнее отличие особей различных поколений одного вида, существующих в разные сезоны. Например,
- 20. Экспрессия гена - реализация наследственной информ., закодированной в гене, в функциональный продукт- РНК или белок. Существуют
- 21. Комбинативная изменчивость - изменчивость связанная с процессами происходящими при мейозе и при слиянии двух отличающихся друг
- 22. Мутационная изменчивость - это вновь возникающие изменения в наследственных структурах клетки под воздействием факторов внешней или
- 23. Мутации - это изменения генотипа, происходящие под влиянием факторов внешней и внутренней среды. - редкие, случайно
- 24. Мутации Генная (изменение структуры гена) изменение ДНК - нарушение порядка нуклеотидов Геномные (изменение количества хромосом в
- 25. Мобильные генетические элементы – транспозоны, ретротранспозоны. Транспозиционная активность МГЭ является основной причиной возникновения спонтанных мутаций. МГЭ
- 27. КЛАССИФИКАЦИЯ МУТАЦИЙ
- 28. КЛАССИФИКАЦИЯ МУТАЦИЙ
- 29. КЛАССИФИКАЦИЯ МУТАЦИЙ
- 30. КЛАССИФИКАЦИЯ МУТАЦИЙ
- 31. Мутагены – факторы, вызывающие стойкие наследственные изменения в организме.
- 32. Основные характеристики мутационной изменчивости 1. Мутационные изменения возникают внезапно, и в результате у организма появляются новые
- 33. Классификация моногенных синдромов Так как проявление моногенных болезней зависит от природы мутантного гена, существует их классификация
- 34. Аутосомно-доминантный тип наследования характеризуется следующими признаками: больные имеются в каждом поколении; больной ребенок у больных родителей;
- 35. Аутосомно-доминантный тип наследования Брахидактилия – короткопалость, которая выражается в укорочении фаланговых костей на пальцах. Существует целая
- 36. Брахидактилия Причиной заболевания являются гетерозиготные мутации в гене тирозин-киназного рецептора ROR2. Установлено, что этот ген экспрессируется
- 37. Робинов синдром Гомозиготные (доминантные и рецессивные) мутации в гене ROR2 обуславливают Робинов синдром, характеризующийся поражением конечностей
- 38. Аутосомно-доминантный тип наследования Ретинобластома — злокачественная опухоль эмбриональной сетчатки глаза. Встречается примерно у 1 новорожденного на
- 39. Ретинобластома
- 40. Ретинобластома если ребёнок наследует мутантный аллель гена Rb, то вторая мутация, происходящая уже в ретинобласте, ведёт
- 41. Аутосомно-доминантный тип наследования Нейрофиброматоз - тяжелая многосистемная болезнь. Популяционная частота - 1:3500 новорожденных. Ген картирован -
- 42. Нейрофиброматоз Чаще наблюдается нейрофиброматоз I типа (болезнь Реклингаузена). В этом случае имеются доминантные мутации в гене
- 43. Аутосомно-доминантный тип наследования Ахондроплазия (хондродистрофия) – диспропорциональная карликовость. У больных нарушаются рост и развитие хрящевой ткани
- 44. Ахондроплазия ахондроплазия вызывается мутацией в гене, кодирующем белок FGFR3 (рецептор 3 к фактору роста фибробластов). При
- 45. Аутосомно-доминантный тип наследования Синдром Марфана (архнодактилия) болезнь, причиной которой является мутация гена белка фибриллина (15q21). Популяционная
- 46. Аутосомно-рецессивный тип наследования характеризуется следующими признаками: больные не в каждом поколении; больной ребенок (гомозигота) рождается у
- 47. Альбинизм врожденное отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза встречается в разных популяциях с
- 48. Альбинизм у представителей разных рас (из ru.wikipedia.org)
- 49. Альбинизм человека
- 50. Типы альбинизма Глазокожный альбинизм 1 А - самая тяжелая форма альбинизма. Он появляется в результате миссенс,
- 51. Фенилкетонурия (ФКУ) встречается с частотой 1:6000 - 1:10 000. Вызвана мутацией гена, который отвечает за синтез
- 52. Фенилкетонурия Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает
- 53. Врожденные нарушения метаболизма фенилаланина и тирозина
- 54. Прогерия Больные прогерией часто имеют характерный внешний вид: низкий рост, относительно большая голова и уменьшенная лицевая
- 55. Прогерия характеризуется комплексом изменений кожи и внутренних органов, обусловленных преждевременным старением организма. Основными формами является детская
- 56. Детская прогерия Причина детской прогерии — мутации гена LMNA, кодирующего ламин А (1q22). Вероятный тип наследования:
- 57. Детская прогерия клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется рост ребенка, отмечаются атрофические
- 58. Детская прогерия Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство источников указывают возраст смерти
- 59. синдром Вернера Прогерия взрослых имеет аутосомно-рецессивный тип наследования (мутации в гене WRN, локализованном в хромосоме 8p12-p11.2).
- 60. ДНК-геликазы расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических процессов: репликация ДНК, транскрипция РНК
- 61. синдром Вернера Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост, гипогонадизм. Обычно на третьем
- 62. Синдром Блума Обусловлен мутациями в гене BLM, принадлежащем к генам ДНК-геликаз. Тип наследования – аутосомно-рецессивный (19q13.3).
- 63. Ксеродерма пигментная Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету (ультрафиолету), что проявляется в
- 64. Ксеродерма пигментная заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов. Существует семь комплементарных групп генов
- 65. Типы пигментной ксеродермы
- 66. Анемия Фанкони (Fanconi anemia) Развивается у детей в возрасте от 4 до 10 лет. Характеризуются аплазией
- 67. Анемия Фанкони Всего известно 7 генов, способных приводить к анемии Фанкони: FancA, FancB, FancC, FancD, FancE,
- 68. Анемия Фанкони Анемия Фанкони как и предыдущие заболевания (с. Блума, пигментная ксеродерма, атаксия-телангиэктазия) относятся к разряду
- 69. Роль теломер в старении Во многих клетках человека утрата способности клеток к делению связана с утратой
- 70. Общие причины синдромов преждевременного старения Нарушение свойств теломер, хроматина и клеточного ядра Нарушение репарации и репликации
- 71. Х-сцепленный рецессивный тип наследования характеризуется следующими признаками: больные появляются не в каждом поколении; больной ребенок рождается
- 72. Дальтонизм - частичная цветовая слепота, один из видов нарушения цветового зрения. Д. впервые описан в 1794
- 74. Гемофилия А - тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается с частотой 1:2500 новорожденных
- 75. Миодистрофия Дюшенна - тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в плазме крови креатинфосфокиназы. Фенотипически
- 76. Мышечная дистрофия Дюшенна: псевдогипертрофия икроножных мышц, ряд последовательных движений при принятии вертикального положения (симптом «лестницы»).
- 77. Х-сцепленный доминантный тип наследования сходен с аутосомно-доминантным, за исключением того, что мужчина передает этот признак только
- 78. Рахит, резистентный к витамину D Происходит нарушение многих видов обмена веществ, что приводит к нарушению костеобразования
- 79. Синдром Марфана
- 80. Увеличение живота у ребенка с гликогенозом. На поздней стадии развития наблюдаются множественные деформации скелета
- 81. а - новорожденная девочка с двойственным строением наружных половых органов; б — девочка 1,5 лет с
- 82. Синдром Эдвардса
- 83. Кисть больного с синдромом трисомии 18 (характерное расположение пальцев).
- 84. Дети с синдромом Дауна. А - европеоид, Б - негроид, В - представитель азиатской расы. Общие
- 85. Внешний вид больной с синдромом Шерешевского—Тернера (выраженный шейный птеригиум; широкая грудная клетка; соски молочных желез гипопластичны,
- 86. Болезнь Шерешевского — Тернера. Особенности телосложения
- 87. Внешний вид больного с синдромом Клайнфельтера (высокий рост, непропорционально длинные конечности, феминизированное телосложение) (47, XXY).
- 88. Синдром тетрасомии X (48, ХХХХ).
- 89. Внешний вид больного с синдромом трисомии по 13 хромосоме (синдром Орбели): а – аномалии лица; б
- 91. Скачать презентацию



Изменчивость
свойство живых организмов приобретать в процессе индивидуального развития новые признаки
Изменчивость
свойство живых организмов приобретать в процессе индивидуального развития новые признаки

Формы изменчивости организмов
Формы изменчивости организмов
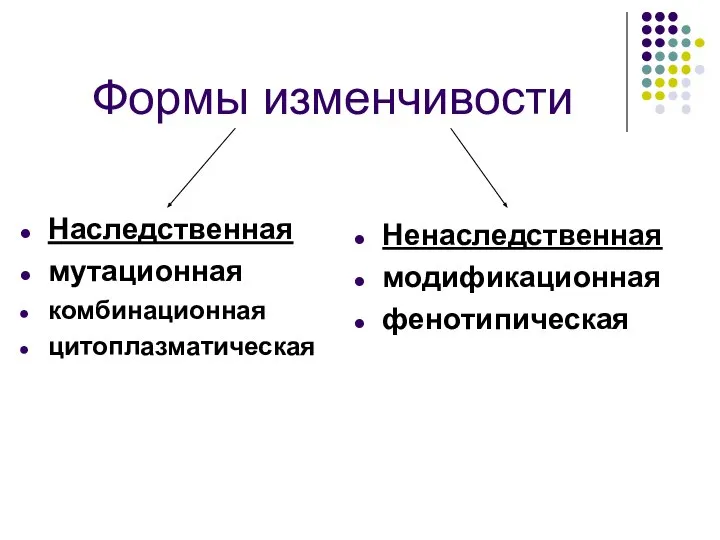
Формы изменчивости
Наследственная
мутационная
комбинационная
цитоплазматическая
Ненаследственная
модификационная
фенотипическая
Формы изменчивости
Наследственная
мутационная
комбинационная
цитоплазматическая
Ненаследственная
модификационная
фенотипическая
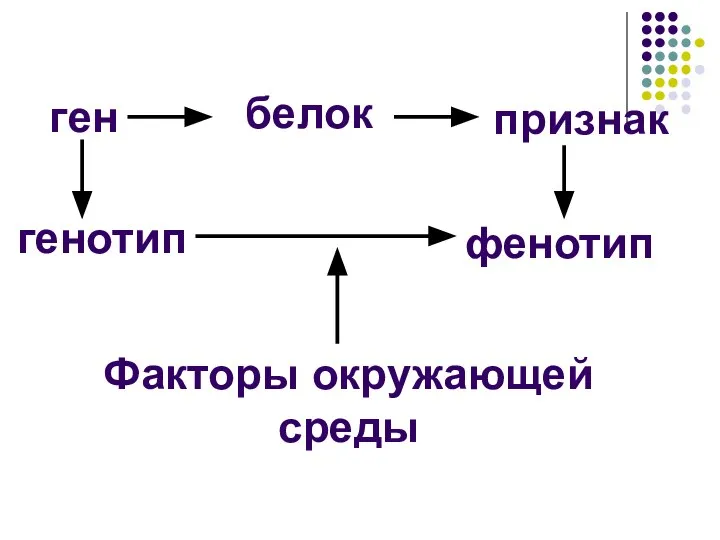
ген
белок
признак
генотип
фенотип
Факторы окружающей среды
ген
белок
признак
генотип
фенотип
Факторы окружающей среды

Изменчивость организма, возникающая под влиянием факторов внешней среды и не затрагивающая
Изменчивость организма, возникающая под влиянием факторов внешней среды и не затрагивающая
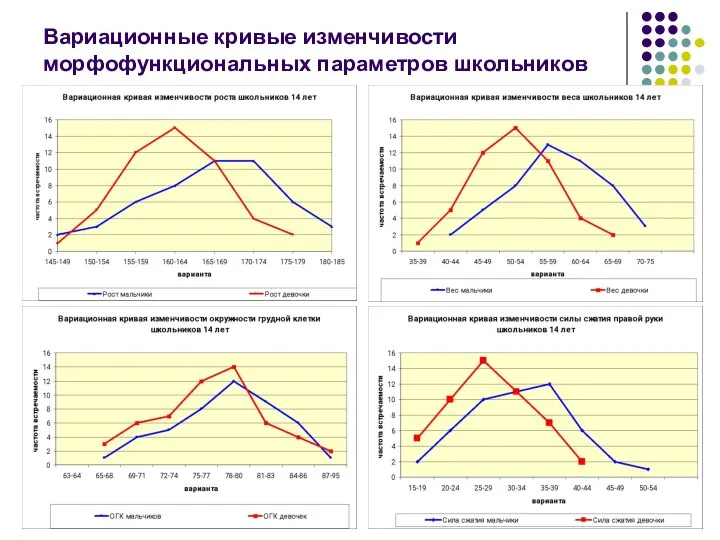
Вариационные кривые изменчивости морфофункциональных параметров школьников
Вариационные кривые изменчивости морфофункциональных параметров школьников

Норма реакции – степень варьирования признака или пределы модификационной изменчивости, обусловленные
Норма реакции – степень варьирования признака или пределы модификационной изменчивости, обусловленные

Выводы:
1. Проявление признака не выходит за пределы нормы реакции, которая
Выводы:
1. Проявление признака не выходит за пределы нормы реакции, которая

Основные характеристики модификационной изменчивости
1. Зависит от окружающих условий.
2. Носит групповой
Основные характеристики модификационной изменчивости
1. Зависит от окружающих условий.
2. Носит групповой

Средняя величина признака
V1 х p1 + V2 х p2+ Vn
Средняя величина признака
V1 х p1 + V2 х p2+ Vn

Наследственная (генотипическая) изменчивость
Комбинативная
Мутационная
Генная
Хромосомная
Геномная
Наследственная (генотипическая) изменчивость
Комбинативная
Мутационная
Генная
Хромосомная
Геномная

Морфоз (случайная фенотипическая изменчивость) – необратимые изменения фенотипа, которые человек получает
Морфоз (случайная фенотипическая изменчивость) – необратимые изменения фенотипа, которые человек получает

Характкристики:
1. Не имеет приспособительного характера;
2. Возникает под действием неблагоприятных
Характкристики:
1. Не имеет приспособительного характера;
2. Возникает под действием неблагоприятных

Пенетрантность – процент реализации гена в признак. Пенетрантность выражается отношением числа
Пенетрантность – процент реализации гена в признак. Пенетрантность выражается отношением числа

Фенокопия — ненаследуемое изменение фенотипа, которое возникает под влиянием внешних факторов и
Фенокопия — ненаследуемое изменение фенотипа, которое возникает под влиянием внешних факторов и

Сезонная изменчивость — это внешнее отличие особей различных поколений одного вида, существующих
Сезонная изменчивость — это внешнее отличие особей различных поколений одного вида, существующих

Экспрессия гена - реализация наследственной информ., закодированной в гене, в функциональный
Экспрессия гена - реализация наследственной информ., закодированной в гене, в функциональный

Комбинативная изменчивость
- изменчивость связанная с процессами происходящими при мейозе и
Комбинативная изменчивость
- изменчивость связанная с процессами происходящими при мейозе и

Мутационная изменчивость
- это вновь возникающие изменения в наследственных структурах
Мутационная изменчивость
- это вновь возникающие изменения в наследственных структурах

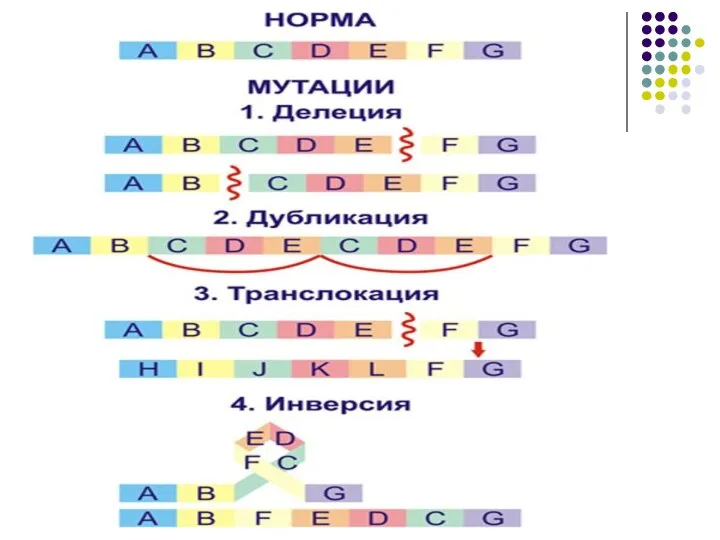
Мутации
- это изменения генотипа, происходящие под влиянием факторов внешней и
Мутации
- это изменения генотипа, происходящие под влиянием факторов внешней и
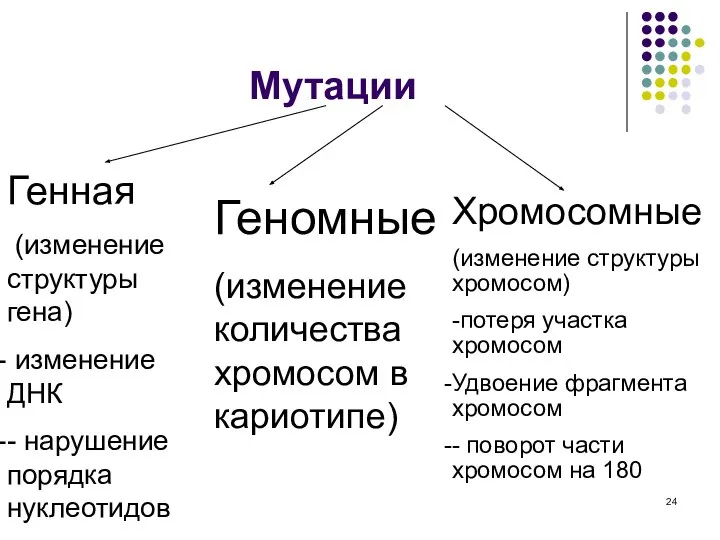
Мутации
Генная
(изменение структуры гена)
изменение ДНК
- нарушение порядка нуклеотидов
Геномные
(изменение
Мутации
Генная
(изменение структуры гена)
изменение ДНК
- нарушение порядка нуклеотидов
Геномные
(изменение

Мобильные генетические элементы – транспозоны, ретротранспозоны.
Транспозиционная активность МГЭ является основной причиной
Мобильные генетические элементы – транспозоны, ретротранспозоны.
Транспозиционная активность МГЭ является основной причиной
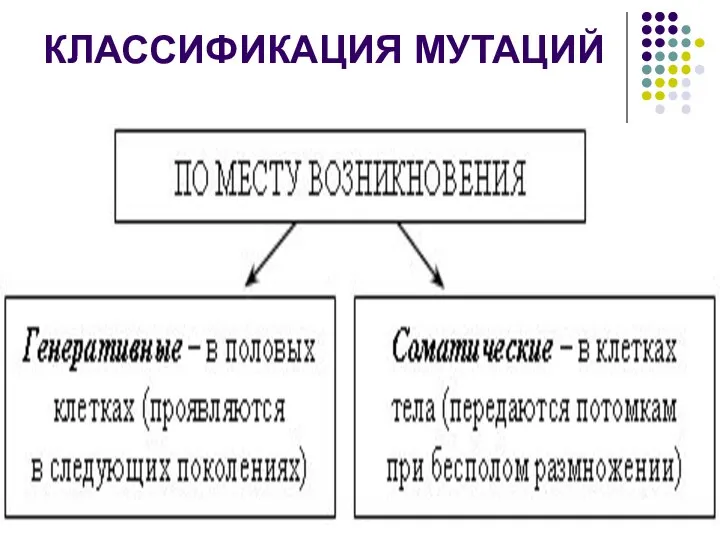
КЛАССИФИКАЦИЯ МУТАЦИЙ
КЛАССИФИКАЦИЯ МУТАЦИЙ
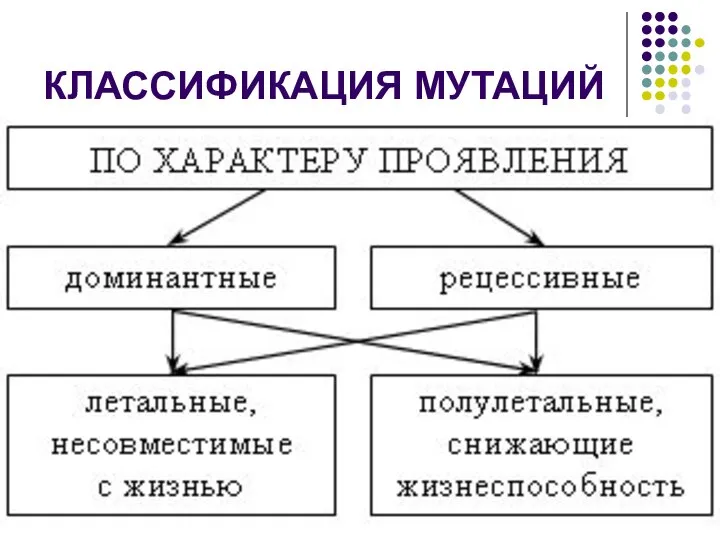
КЛАССИФИКАЦИЯ МУТАЦИЙ
КЛАССИФИКАЦИЯ МУТАЦИЙ
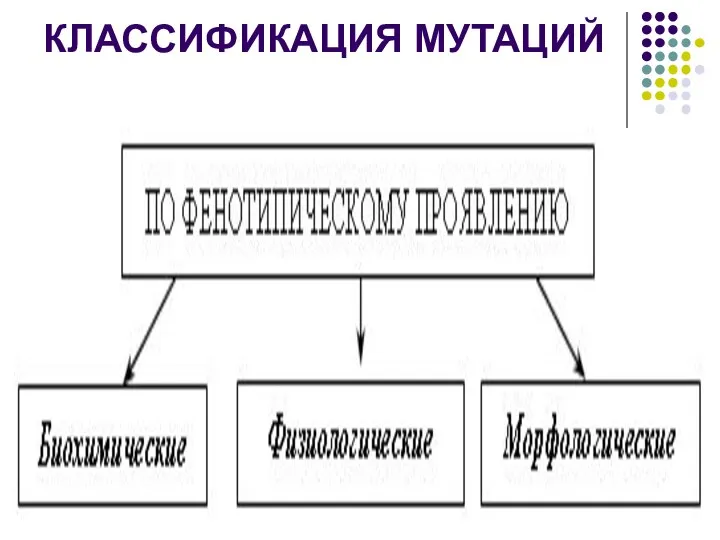
КЛАССИФИКАЦИЯ МУТАЦИЙ
КЛАССИФИКАЦИЯ МУТАЦИЙ
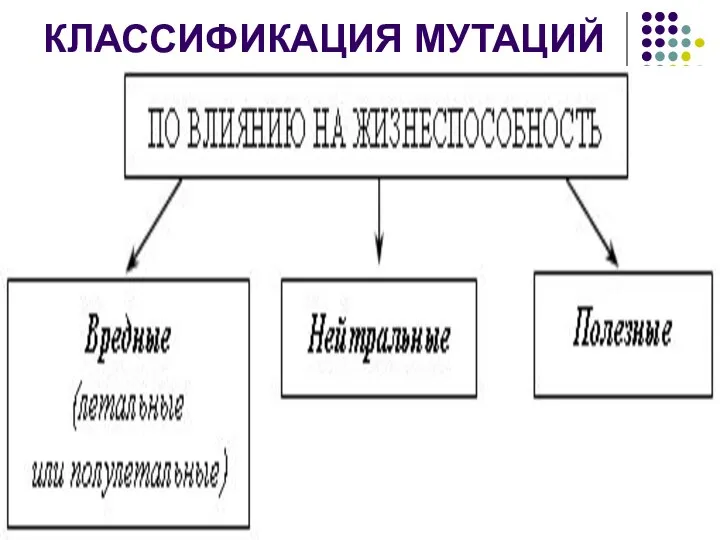
КЛАССИФИКАЦИЯ МУТАЦИЙ
КЛАССИФИКАЦИЯ МУТАЦИЙ
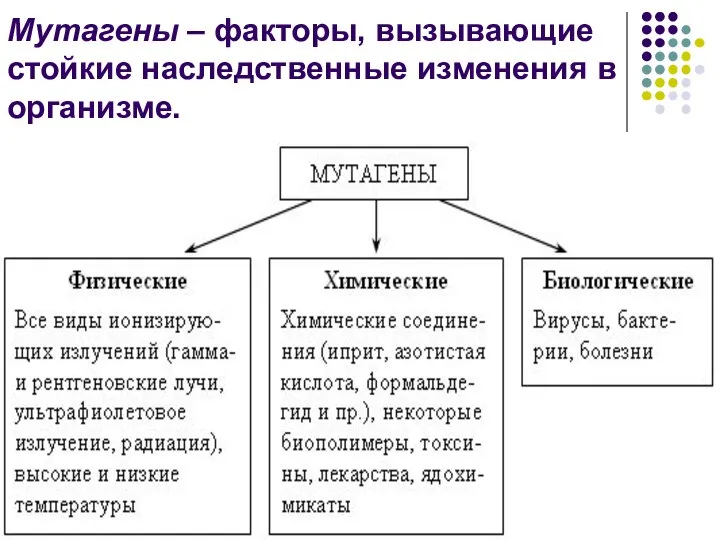
Мутагены – факторы, вызывающие стойкие наследственные изменения в организме.
Мутагены – факторы, вызывающие стойкие наследственные изменения в организме.

Основные характеристики мутационной изменчивости
1. Мутационные изменения возникают внезапно, и в результате
Основные характеристики мутационной изменчивости
1. Мутационные изменения возникают внезапно, и в результате

Классификация моногенных синдромов
Так как проявление моногенных болезней зависит от природы мутантного
Классификация моногенных синдромов
Так как проявление моногенных болезней зависит от природы мутантного

Аутосомно-доминантный тип наследования
характеризуется следующими признаками:
больные имеются в каждом
Аутосомно-доминантный тип наследования
характеризуется следующими признаками:
больные имеются в каждом
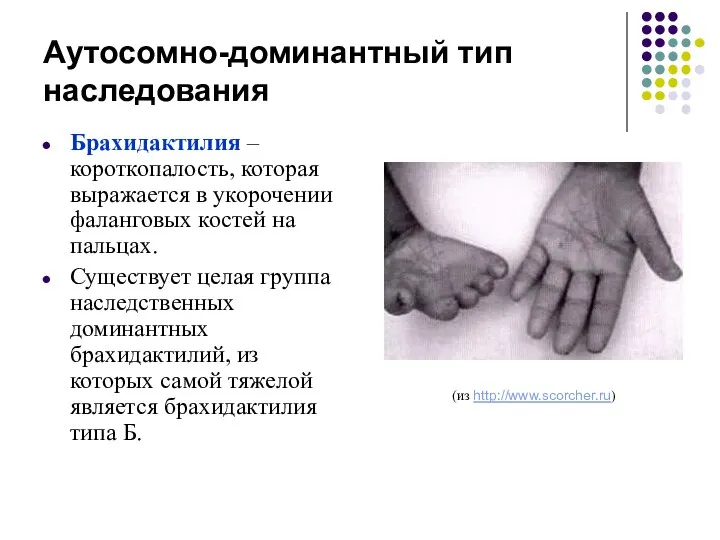
Аутосомно-доминантный тип наследования
Брахидактилия – короткопалость, которая выражается в укорочении фаланговых костей
Аутосомно-доминантный тип наследования
Брахидактилия – короткопалость, которая выражается в укорочении фаланговых костей

Брахидактилия
Причиной заболевания являются гетерозиготные мутации в гене тирозин-киназного рецептора ROR2.
Установлено, что
Брахидактилия
Причиной заболевания являются гетерозиготные мутации в гене тирозин-киназного рецептора ROR2.
Установлено, что
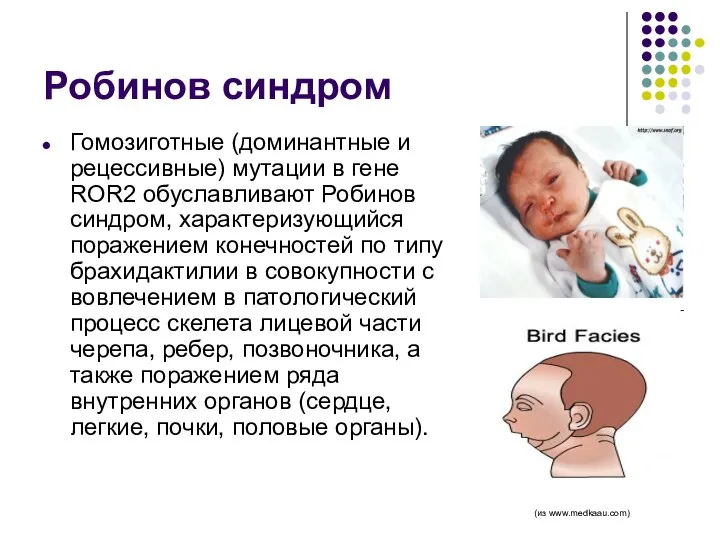
Робинов синдром
Гомозиготные (доминантные и рецессивные) мутации в гене ROR2 обуславливают Робинов
Робинов синдром
Гомозиготные (доминантные и рецессивные) мутации в гене ROR2 обуславливают Робинов
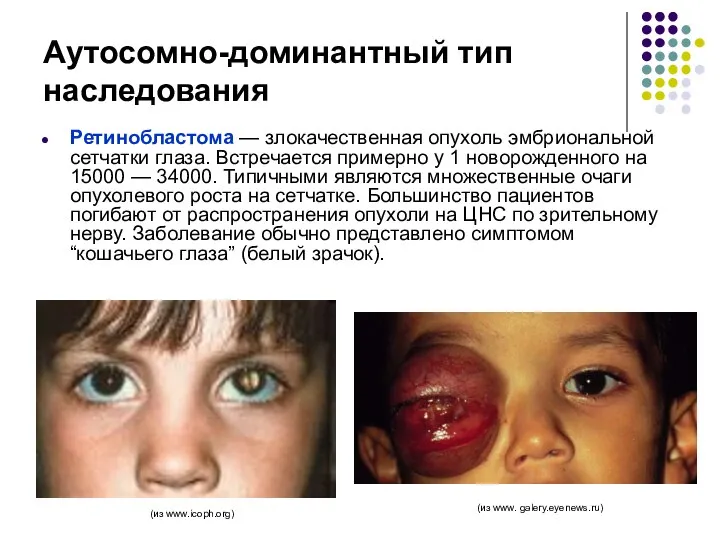
Аутосомно-доминантный тип наследования
Ретинобластома — злокачественная опухоль эмбриональной сетчатки глаза. Встречается примерно
Аутосомно-доминантный тип наследования
Ретинобластома — злокачественная опухоль эмбриональной сетчатки глаза. Встречается примерно

Ретинобластома
Ретинобластома

Ретинобластома
если ребёнок наследует мутантный аллель гена Rb, то вторая мутация, происходящая
Ретинобластома
если ребёнок наследует мутантный аллель гена Rb, то вторая мутация, происходящая
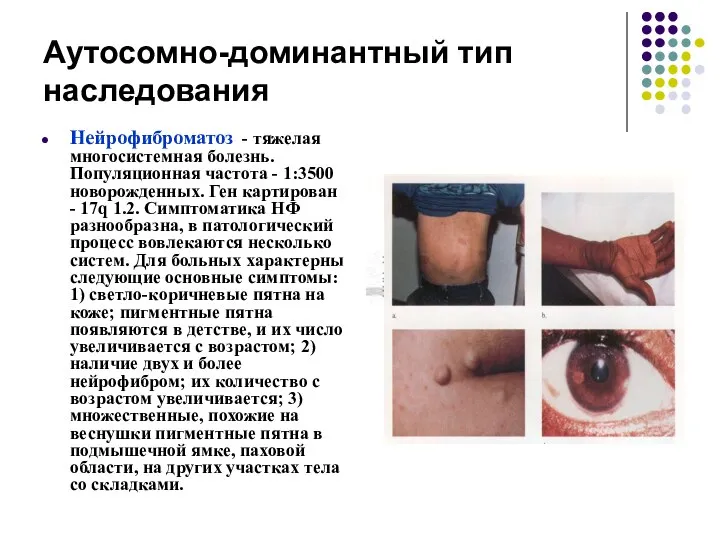
Аутосомно-доминантный тип наследования
Нейрофиброматоз - тяжелая многосистемная болезнь. Популяционная частота - 1:3500
Аутосомно-доминантный тип наследования
Нейрофиброматоз - тяжелая многосистемная болезнь. Популяционная частота - 1:3500

Нейрофиброматоз
Чаще наблюдается нейрофиброматоз I типа (болезнь Реклингаузена). В этом случае имеются
Нейрофиброматоз
Чаще наблюдается нейрофиброматоз I типа (болезнь Реклингаузена). В этом случае имеются
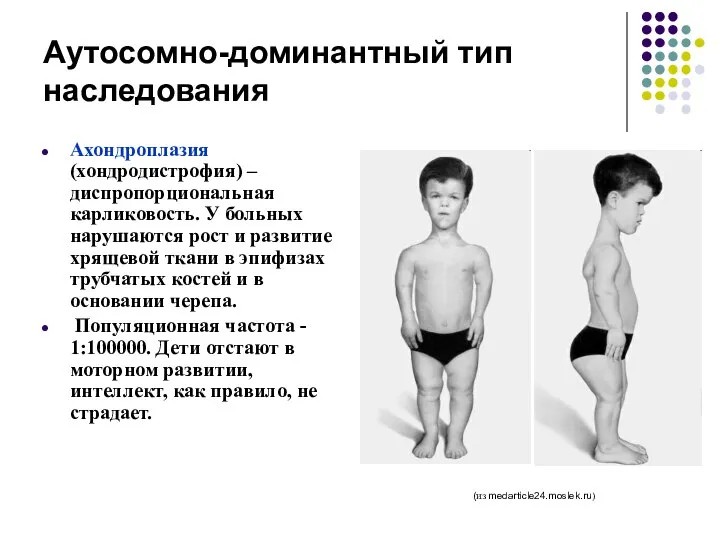
Аутосомно-доминантный тип наследования
Ахондроплазия (хондродистрофия) – диспропорциональная карликовость. У больных нарушаются рост
Аутосомно-доминантный тип наследования
Ахондроплазия (хондродистрофия) – диспропорциональная карликовость. У больных нарушаются рост

Ахондроплазия
ахондроплазия вызывается мутацией в гене, кодирующем белок FGFR3 (рецептор 3 к
Ахондроплазия
ахондроплазия вызывается мутацией в гене, кодирующем белок FGFR3 (рецептор 3 к
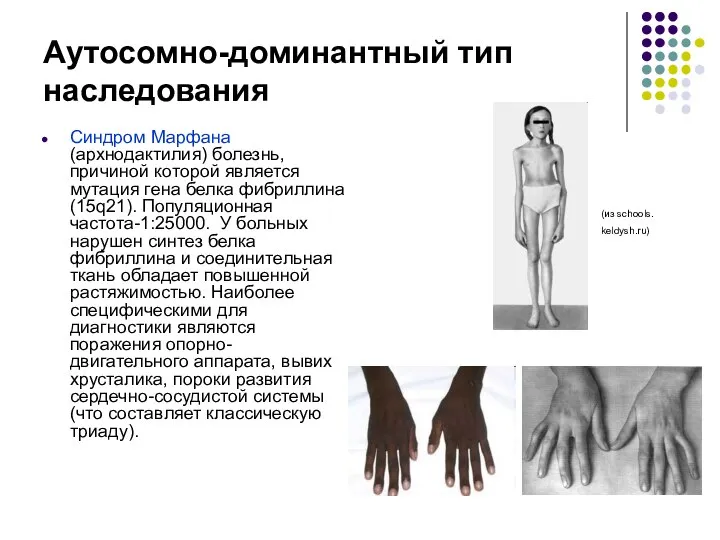
Аутосомно-доминантный тип наследования
Синдром Марфана (архнодактилия) болезнь, причиной которой является мутация гена
Аутосомно-доминантный тип наследования
Синдром Марфана (архнодактилия) болезнь, причиной которой является мутация гена

Аутосомно-рецессивный тип наследования
характеризуется следующими признаками:
больные не в каждом
Аутосомно-рецессивный тип наследования
характеризуется следующими признаками:
больные не в каждом

Альбинизм
врожденное отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза встречается в
Альбинизм
врожденное отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза встречается в

Альбинизм у представителей разных рас
(из ru.wikipedia.org)
Альбинизм у представителей разных рас
(из ru.wikipedia.org)
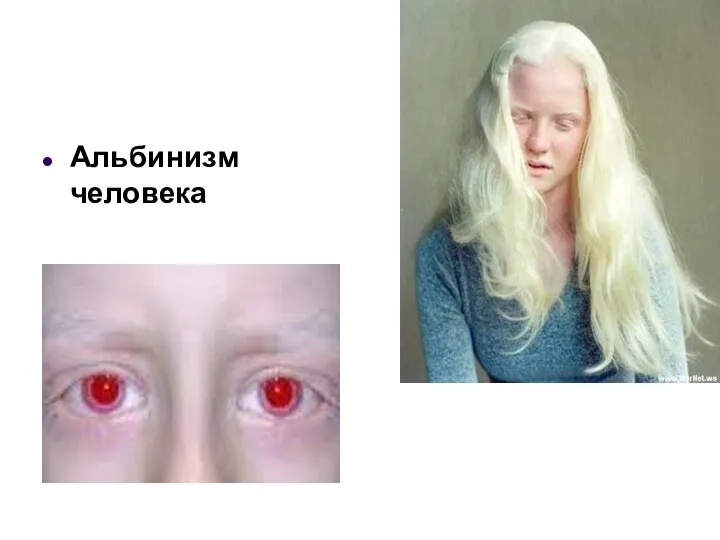
Альбинизм человека
Альбинизм человека

Типы альбинизма
Глазокожный альбинизм 1 А - самая тяжелая форма альбинизма. Он
Типы альбинизма
Глазокожный альбинизм 1 А - самая тяжелая форма альбинизма. Он
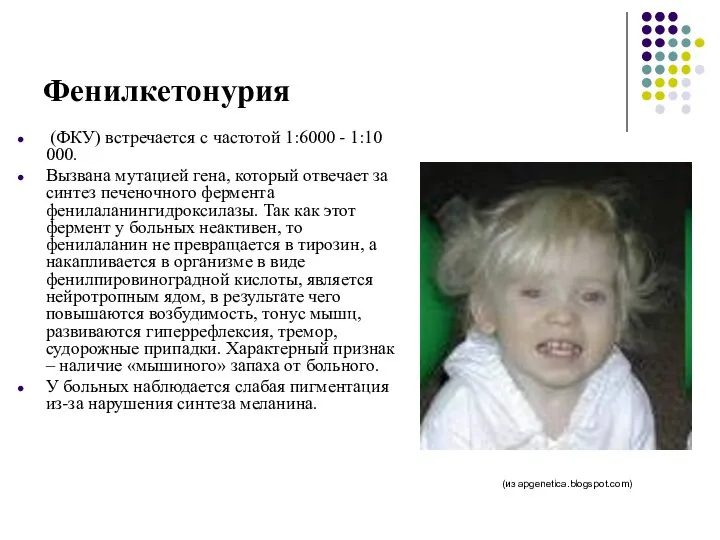
Фенилкетонурия
(ФКУ) встречается с частотой 1:6000 - 1:10 000.
Вызвана мутацией
Фенилкетонурия
(ФКУ) встречается с частотой 1:6000 - 1:10 000.
Вызвана мутацией

Фенилкетонурия
Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и
Фенилкетонурия
Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и
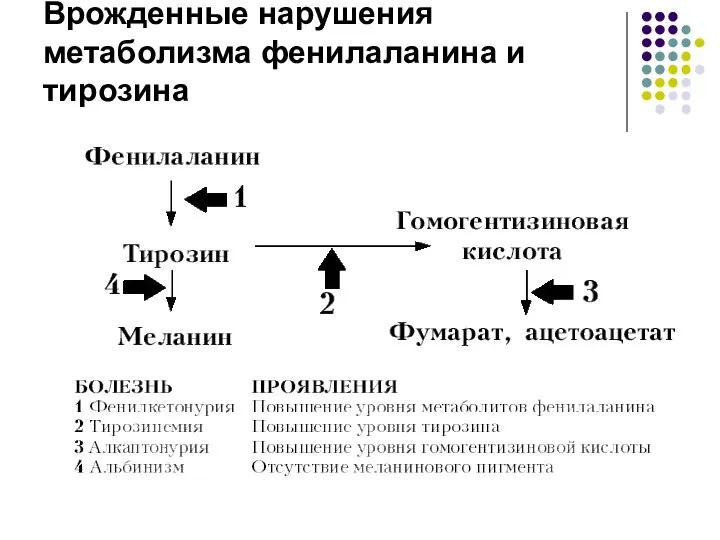
Врожденные нарушения метаболизма фенилаланина и тирозина
Врожденные нарушения метаболизма фенилаланина и тирозина
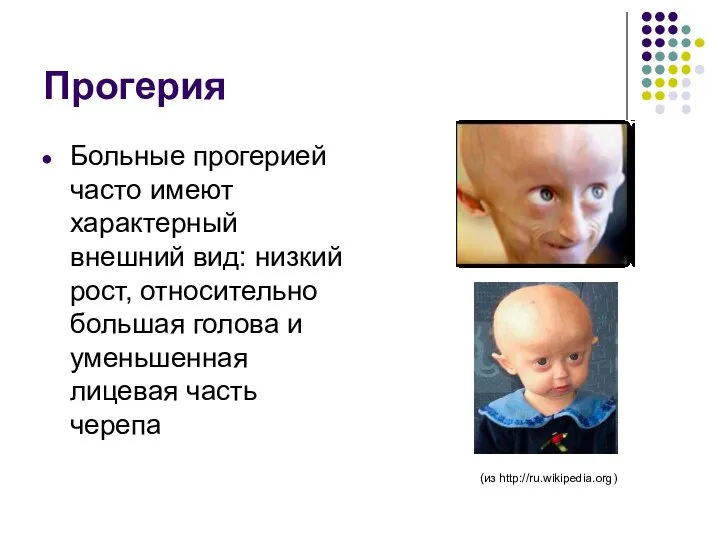
Прогерия
Больные прогерией часто имеют характерный внешний вид: низкий рост, относительно большая
Прогерия
Больные прогерией часто имеют характерный внешний вид: низкий рост, относительно большая

Прогерия
характеризуется комплексом изменений кожи и внутренних органов, обусловленных преждевременным старением организма.
Прогерия
характеризуется комплексом изменений кожи и внутренних органов, обусловленных преждевременным старением организма.

Детская прогерия
Причина детской прогерии — мутации гена LMNA, кодирующего ламин А
Детская прогерия
Причина детской прогерии — мутации гена LMNA, кодирующего ламин А

Детская прогерия
клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется
Детская прогерия
клинические признаки проявляются обычно на 2—3-м году жизни. Резко замедляется

Детская прогерия
Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство
Детская прогерия
Средняя продолжительность жизни при детской прогерии — 13 лет. Большинство
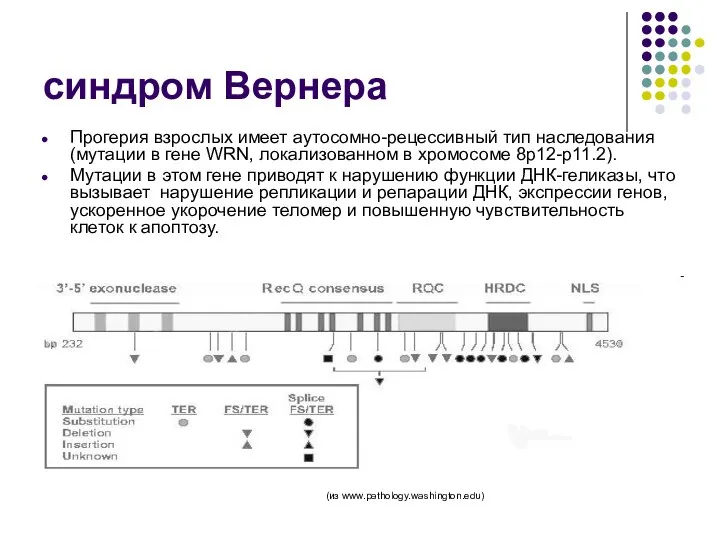
синдром Вернера
Прогерия взрослых имеет аутосомно-рецессивный тип наследования (мутации в гене WRN,
синдром Вернера
Прогерия взрослых имеет аутосомно-рецессивный тип наследования (мутации в гене WRN,
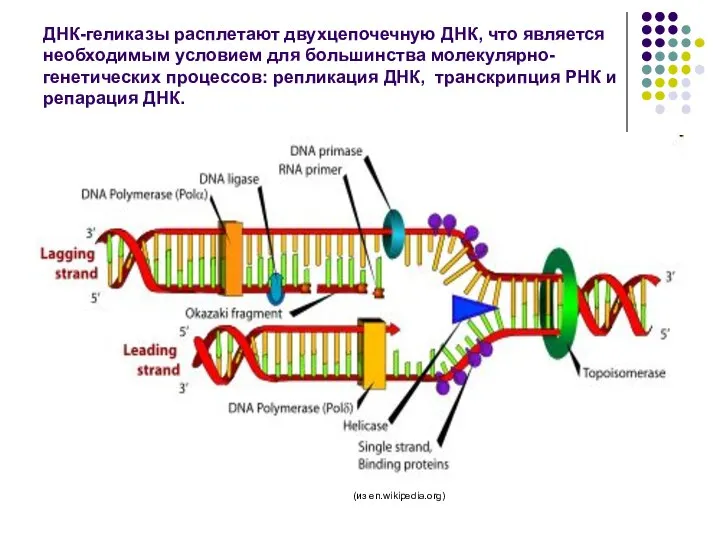
ДНК-геликазы расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических
ДНК-геликазы расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических

синдром Вернера
Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост,
синдром Вернера
Клинически заболевание проявляется в период полового созревания. Отмечаются замедленный рост,
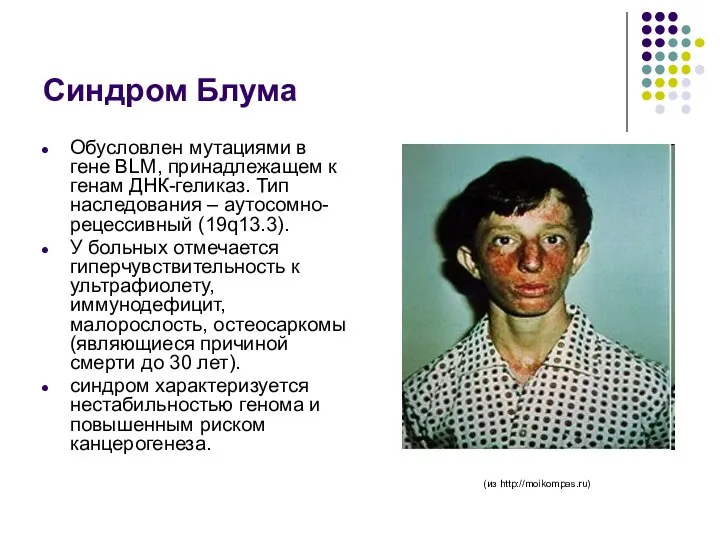
Синдром Блума
Обусловлен мутациями в гене BLM, принадлежащем к генам ДНК-геликаз. Тип
Синдром Блума
Обусловлен мутациями в гене BLM, принадлежащем к генам ДНК-геликаз. Тип
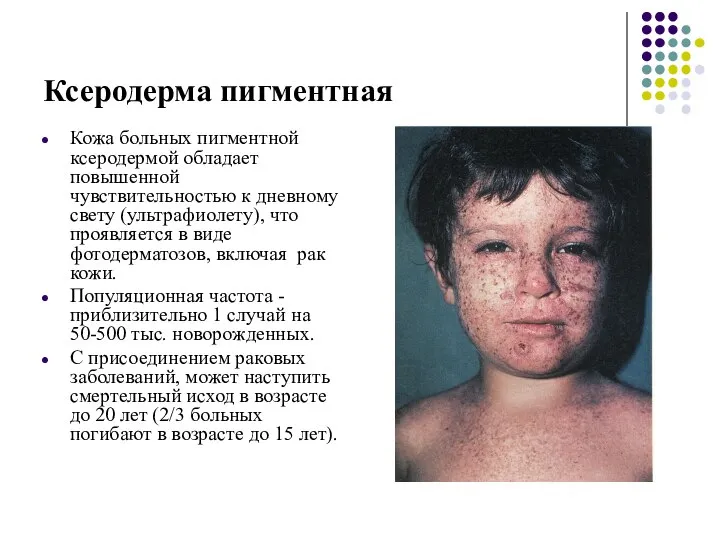
Ксеродерма пигментная
Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету
Ксеродерма пигментная
Кожа больных пигментной ксеродермой обладает повышенной чувствительностью к дневному свету

Ксеродерма пигментная
заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов.
Существует
Ксеродерма пигментная
заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов.
Существует
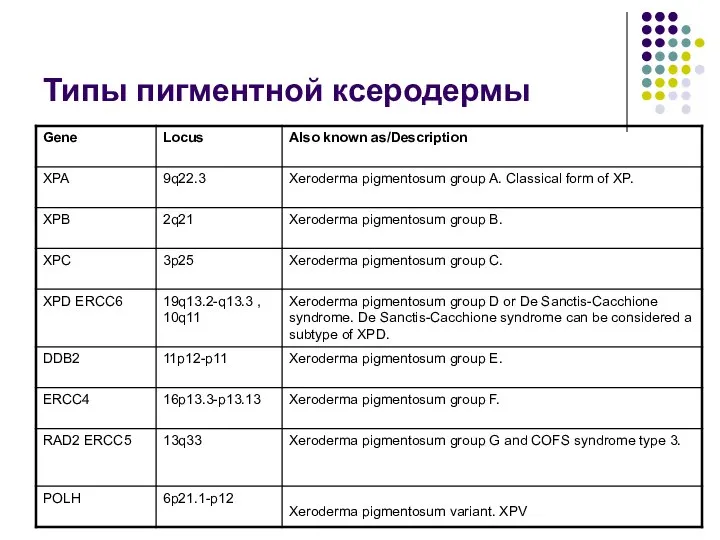
Типы пигментной ксеродермы
Типы пигментной ксеродермы
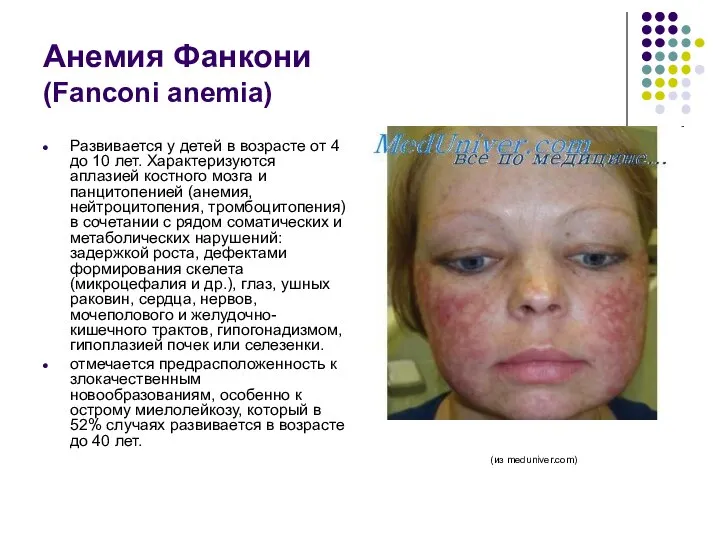
Анемия Фанкони
(Fanconi anemia)
Развивается у детей в возрасте от 4 до
Анемия Фанкони
(Fanconi anemia)
Развивается у детей в возрасте от 4 до
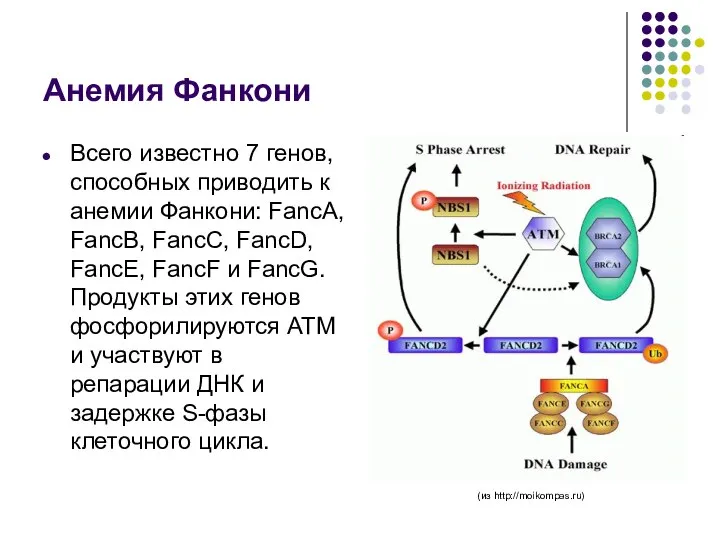
Анемия Фанкони
Всего известно 7 генов, способных приводить к анемии Фанкони: FancA,
Анемия Фанкони
Всего известно 7 генов, способных приводить к анемии Фанкони: FancA,
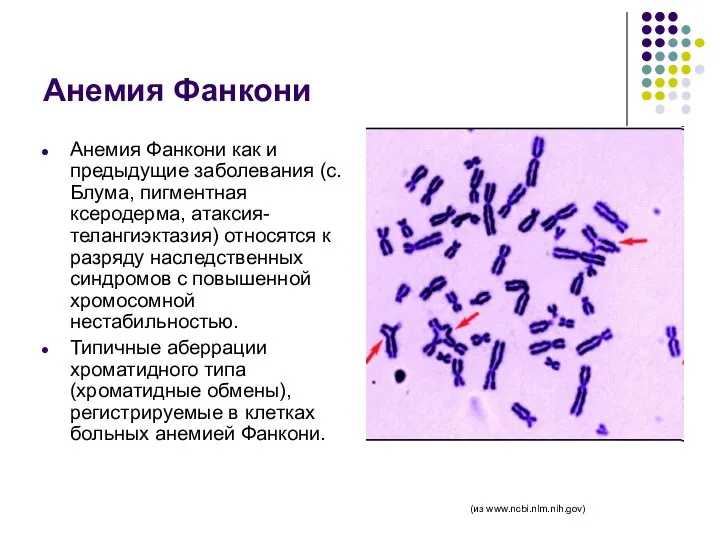
Анемия Фанкони
Анемия Фанкони как и предыдущие заболевания (с. Блума, пигментная ксеродерма,
Анемия Фанкони
Анемия Фанкони как и предыдущие заболевания (с. Блума, пигментная ксеродерма,
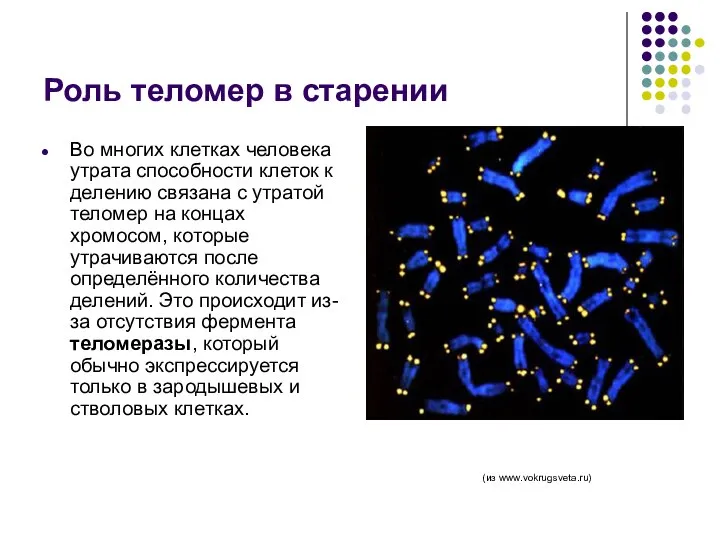
Роль теломер в старении
Во многих клетках человека утрата способности клеток к
Роль теломер в старении
Во многих клетках человека утрата способности клеток к

Общие причины синдромов преждевременного старения
Нарушение свойств теломер, хроматина и клеточного ядра
Общие причины синдромов преждевременного старения
Нарушение свойств теломер, хроматина и клеточного ядра

Х-сцепленный рецессивный тип наследования
характеризуется следующими признаками:
больные появляются не в
Х-сцепленный рецессивный тип наследования
характеризуется следующими признаками:
больные появляются не в

Дальтонизм
- частичная цветовая слепота, один из видов нарушения цветового зрения. Д.
Дальтонизм
- частичная цветовая слепота, один из видов нарушения цветового зрения. Д.


Гемофилия А
- тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается
Гемофилия А
- тяжелое заболевание, обусловленное дефектом фактора VIII свертывания крови. Встречается
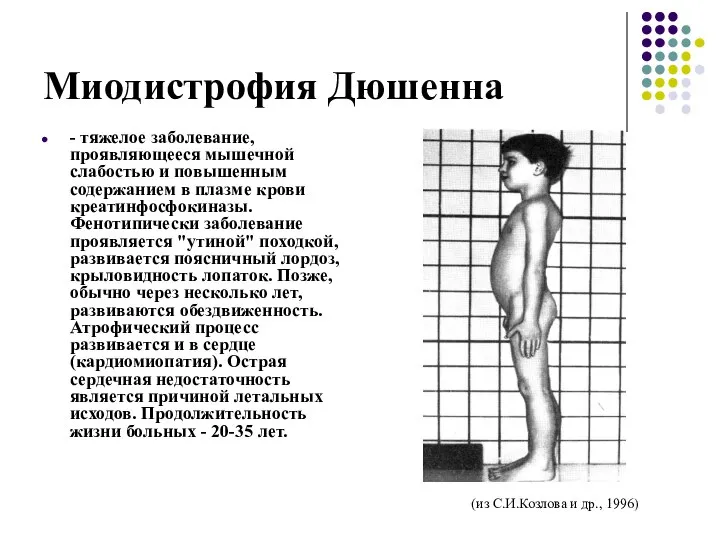
Миодистрофия Дюшенна
- тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в
Миодистрофия Дюшенна
- тяжелое заболевание, проявляющееся мышечной слабостью и повышенным содержанием в
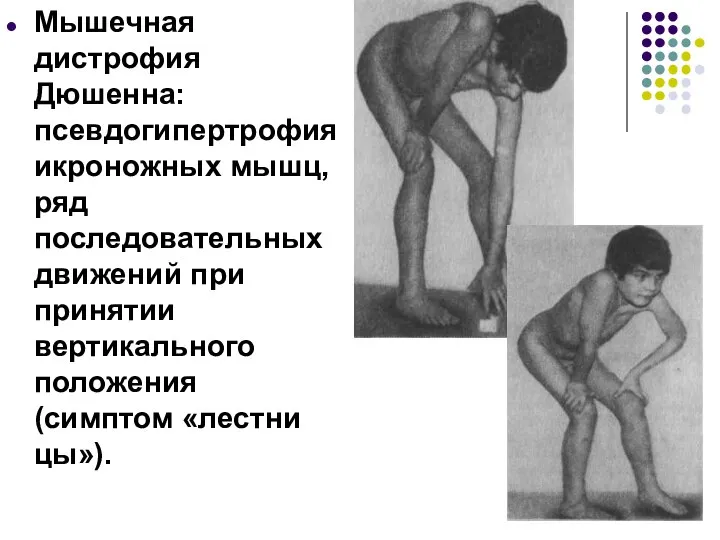
Мышечная дистрофия Дюшенна: псевдогипертрофия икроножных мышц, ряд последовательных движений при принятии
Мышечная дистрофия Дюшенна: псевдогипертрофия икроножных мышц, ряд последовательных движений при принятии

Х-сцепленный доминантный тип наследования
сходен с аутосомно-доминантным, за исключением того, что
Х-сцепленный доминантный тип наследования
сходен с аутосомно-доминантным, за исключением того, что
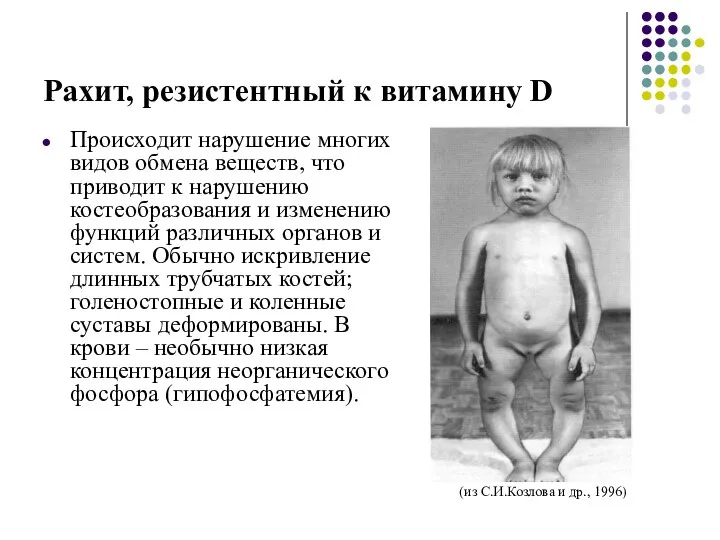
Рахит, резистентный к витамину D
Происходит нарушение многих видов обмена веществ, что
Рахит, резистентный к витамину D
Происходит нарушение многих видов обмена веществ, что
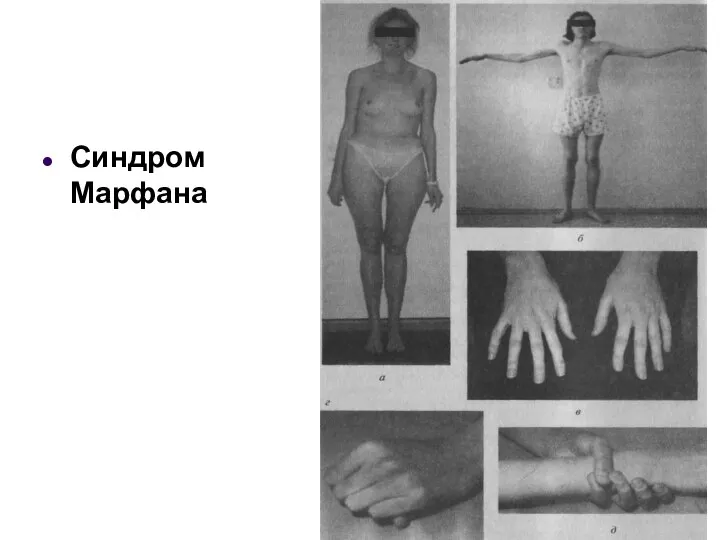
Синдром Марфана
Синдром Марфана

Увеличение живота у ребенка с гликогенозом. На поздней стадии развития наблюдаются
Увеличение живота у ребенка с гликогенозом. На поздней стадии развития наблюдаются
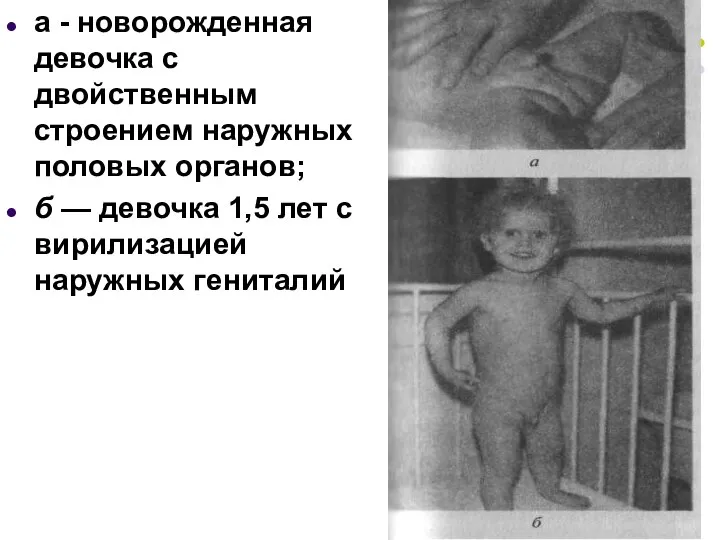
а - новорожденная девочка с двойственным строением наружных половых органов;
б
а - новорожденная девочка с двойственным строением наружных половых органов;
б
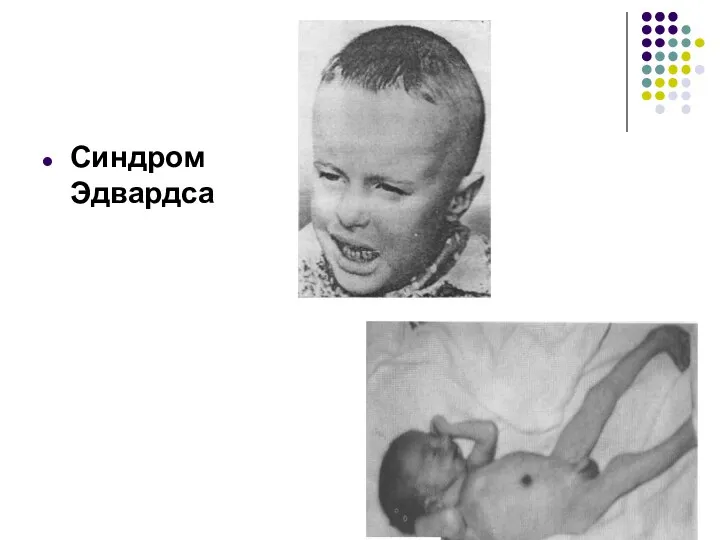
Синдром Эдвардса
Синдром Эдвардса
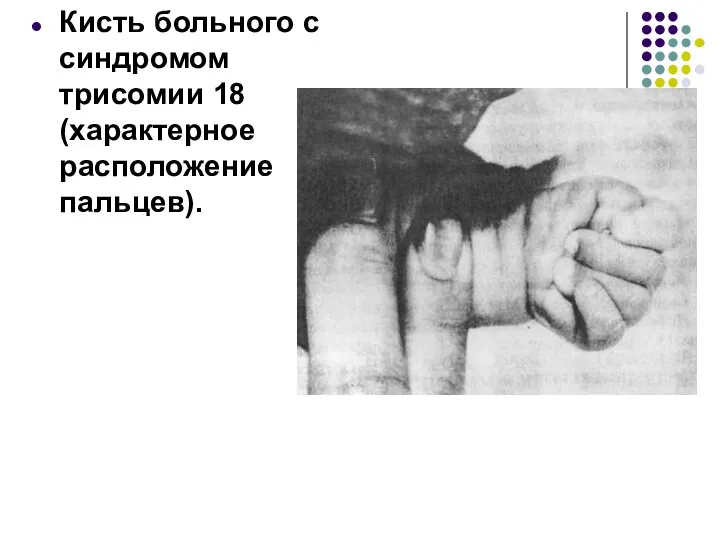
Кисть больного с синдромом трисомии 18 (характерное расположение пальцев).
Кисть больного с синдромом трисомии 18 (характерное расположение пальцев).

Дети с синдромом Дауна. А - европеоид, Б - негроид, В
Дети с синдромом Дауна. А - европеоид, Б - негроид, В

Внешний вид больной с синдромом Шерешевского—Тернера (выраженный шейный птеригиум; широкая грудная
Внешний вид больной с синдромом Шерешевского—Тернера (выраженный шейный птеригиум; широкая грудная
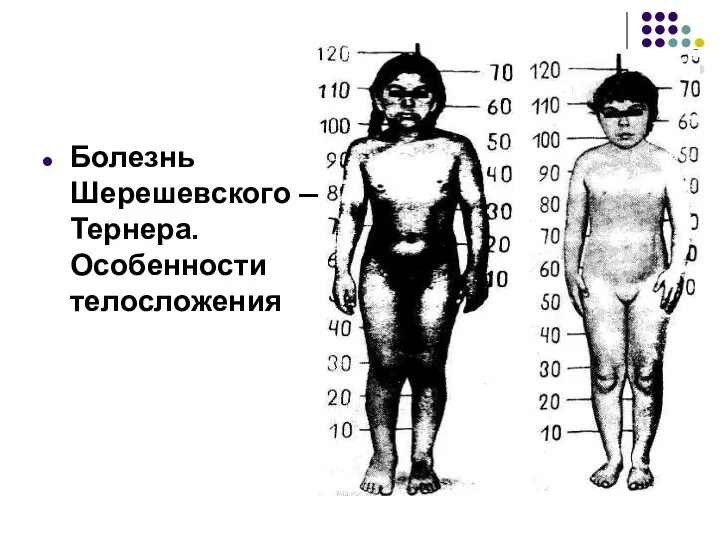
Болезнь Шерешевского — Тернера. Особенности телосложения
Болезнь Шерешевского — Тернера. Особенности телосложения
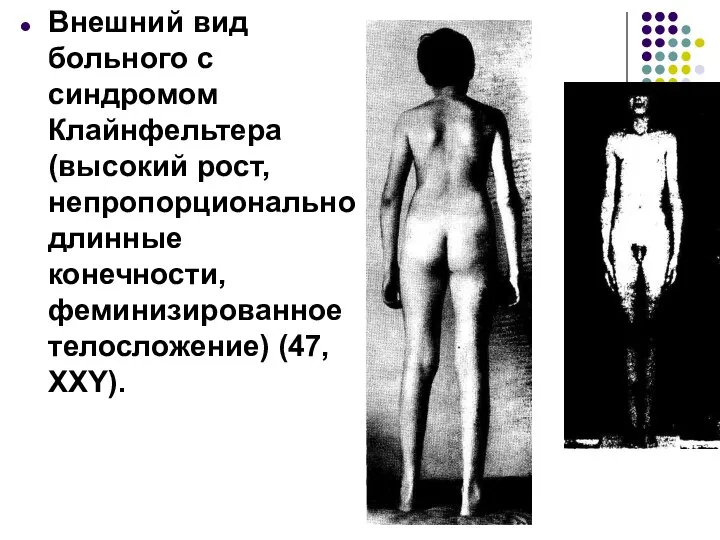
Внешний вид больного с синдромом Клайнфельтера (высокий рост, непропорционально длинные конечности,
Внешний вид больного с синдромом Клайнфельтера (высокий рост, непропорционально длинные конечности,

Синдром тетрасомии X (48, ХХХХ).
Синдром тетрасомии X (48, ХХХХ).
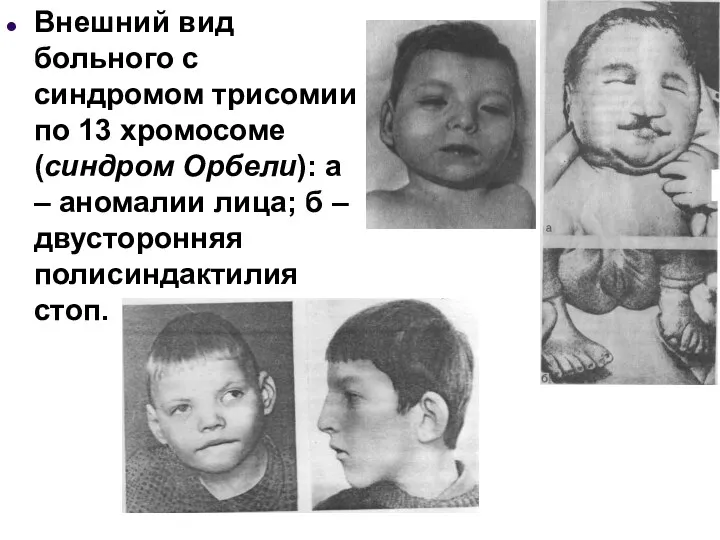
Внешний вид больного с синдромом трисомии по 13 хромосоме (синдром Орбели):
Внешний вид больного с синдромом трисомии по 13 хромосоме (синдром Орбели):
 Нестероидные противовоспалительные препараты
Нестероидные противовоспалительные препараты Заглоточный абсцесс
Заглоточный абсцесс О взаимодействии здравоохранения и Русской Православной Церкви в решении демографических проблем
О взаимодействии здравоохранения и Русской Православной Церкви в решении демографических проблем Основы реабилитации: Инфракрасное излучение
Основы реабилитации: Инфракрасное излучение Изменения в ВНЧС при ортодонтическом вмешательстве
Изменения в ВНЧС при ортодонтическом вмешательстве Қазақстан Республикасының Мемлекеттік фармакопеясы
Қазақстан Республикасының Мемлекеттік фармакопеясы Системная склеродермия
Системная склеродермия Этиология анаэробных раневых, гнойно-воспалительных, и септических инфекций
Этиология анаэробных раневых, гнойно-воспалительных, и септических инфекций Природное и общественное. Отличия человека от животного
Природное и общественное. Отличия человека от животного Реабилитация хирургических больных. Врачебно-трудовая экспертиза. Функции ВКК, МСЭК
Реабилитация хирургических больных. Врачебно-трудовая экспертиза. Функции ВКК, МСЭК Швы хирургические
Швы хирургические Организационно-правовые аспекты первой помощи
Организационно-правовые аспекты первой помощи Осмотр больного неврологического профиля
Осмотр больного неврологического профиля Пищевые токсикоинфекции
Пищевые токсикоинфекции Клинический случай. Лечение зуба 4.5. Кариес дентина
Клинический случай. Лечение зуба 4.5. Кариес дентина Клиническая фармакология препаратов, влияющих на функцию пищеварительной системы
Клиническая фармакология препаратов, влияющих на функцию пищеварительной системы Оценка функционального состояния пациента
Оценка функционального состояния пациента Презентация по медицине Трансплантация органов и тканей
Презентация по медицине Трансплантация органов и тканей  ГБУЗ МО Дмитровская городская больница гинекологическое отделение
ГБУЗ МО Дмитровская городская больница гинекологическое отделение Вакцины. Вакцинация против пневмококковой инфекции
Вакцины. Вакцинация против пневмококковой инфекции Цитомегаловирусты, герпестік, хламидиялық инфекция және жүктілік
Цитомегаловирусты, герпестік, хламидиялық инфекция және жүктілік Акмеология как условие повышения качества образования
Акмеология как условие повышения качества образования Принципы диспансерного наблюдения детей
Принципы диспансерного наблюдения детей Выписка, получение, хранение и учет лекарственных средств
Выписка, получение, хранение и учет лекарственных средств Гормональные средства. Часть 1
Гормональные средства. Часть 1 Скрининг рака шейки матки. Региональная программа Тамбовской области
Скрининг рака шейки матки. Региональная программа Тамбовской области Мужская половая система
Мужская половая система Диагностика и лечение миокардитов
Диагностика и лечение миокардитов