- Первичные иммунодефициты

Содержание
- 2. Дефицит системы фагоцитов 10-12% от общей частоты встречаемости перв.ИД Хронический гранулематоз (в бльшинсве случаев связано с
- 3. Клинические проявление хронического гранулематоза Рецидивирующие бактериальные инфекции Образование в тканях гранулем Проявляется, как правило, в детском
- 4. Синдром Чегиак-Хигаши (Чедиак-Хигаси) Потеря нейтрофилами способности высвобождать лизосомальные ферменты Нарушение хемотаксиса
- 5. Клинические проявления Пиогенные рецидивирующие инфекции Альбинизм (высокая фоточувствительность кожи)
- 6. Дефицит системы комплемента 1% от общего количества перв.ИД Генетические дефекты описаны для всех компонентов комплемента С1q
- 7. Синдром Ди-Джорджи(синдром делеции 22 хромосомы) порок развития первого и второго жаберных карманов – порок развития лицевых
- 8. Синдром Ди Джорджи Аплазия(полный) или гипоплазия (частичный) тимуса Триада -недоразвитие тимуса -отсутствие паращитовидных желез -аномалия развития
- 9. Синдром Ди Джорджи Иммунологические последствия Снижение количества и активности Т-лф Количество В-лф в норме Уровень ат
- 10. Синдром Ди Джорджи Клиника: Рецидивирующие вирусные, паразитарные, вирусные инфекции Судороги Дисморфия лица Другие пороки развития –атрезия
- 11. Больная с синдромом Ди-Джорджи 11 лет • ОРВИ 4-5 раз в год • 10 лет –аутоиммунная
- 12. Синдром Незелофа (лимфоцитарная дисгенезия, нормоплазмоцитарная и нормогаммаглобулинемическая аплазия) Гипоплазия тимуса АТ – в норме
- 13. Синдром Незелофа клиника: замедление роста рецидивирующие инфекции кожи и легких кандидоз лимфаденопатия
- 14. Дефицит В-клеточного звена Агамма(гипогамма)глобулинемия (болезнь Брутона) рецессивный тип наследования Дефицит цитоплазматической тирозинкиназы Нарушение дифференцировки В-лф в
- 15. клиника Определяется на 5-9 месяце жизни Рецидивирующие гнойные инфекции Менингиты Отсутсвие реакции лф.узлов и селезенки на
- 16. ТКИД – основные клинические проявления • раннее начало (обычно между 3-9 месяцами жизни) • Гипоплазия лимфоидной
- 17. ТКИД (швейцарский тип) Х-сцепленный тип Дефект на уровне стволовой клетки нежизнеспособность
- 18. Синдром Вискотта-Олдрича (СВО) - ТКИД В основе лежит мутация гена, который кодирует белок (WASP), отвечающий за
- 19. Комбинированные ИД Синдром Вискотта-Олдрича Нарушение активации Т-h и Т-s ) Врожденный дефект тромбоцитов Нарушение способности макрофагов
- 20. Синдром Вискотта-Олдрича Клиника: Экзема Тромбоцитопения Бактериальные и вирусные инфекции
- 21. Больной с СВО 4-х лет • С 3 месяцев до 3-х лет Тяжелый распространенный атопический дерматит
- 22. Синдром Вискотта-Олдрича
- 23. Синдром Луи-Барра (атаксия-телеангиэктазия) Гипоплазия тимуса Гипоплазия лимф. узлов, селезенки Количественная и функциональная недостаточность Т-лимфоцитов Снижение количества
- 24. Синдром Луи-Барра Клиника: Атаксия Телеангиэктазы (на носу, ушах, конъюктиве) Рецидивирующие инфекции Опухолевые заболевания
- 26. Скачать презентацию

Дефицит системы фагоцитов
10-12% от общей частоты встречаемости перв.ИД
Хронический гранулематоз
(в
Дефицит системы фагоцитов
10-12% от общей частоты встречаемости перв.ИД
Хронический гранулематоз
(в

Клинические проявление хронического гранулематоза
Рецидивирующие бактериальные инфекции
Образование в тканях гранулем
Проявляется, как правило,
Клинические проявление хронического гранулематоза
Рецидивирующие бактериальные инфекции
Образование в тканях гранулем
Проявляется, как правило,

Синдром Чегиак-Хигаши
(Чедиак-Хигаси)
Потеря нейтрофилами способности высвобождать лизосомальные ферменты
Нарушение хемотаксиса
Синдром Чегиак-Хигаши
(Чедиак-Хигаси)
Потеря нейтрофилами способности высвобождать лизосомальные ферменты
Нарушение хемотаксиса

Клинические проявления
Пиогенные рецидивирующие инфекции
Альбинизм (высокая фоточувствительность кожи)
Клинические проявления
Пиогенные рецидивирующие инфекции
Альбинизм (высокая фоточувствительность кожи)

Дефицит системы комплемента
1% от общего количества перв.ИД
Генетические дефекты описаны для всех
Дефицит системы комплемента
1% от общего количества перв.ИД
Генетические дефекты описаны для всех

Синдром Ди-Джорджи(синдром делеции 22 хромосомы)
порок развития первого и второго
Синдром Ди-Джорджи(синдром делеции 22 хромосомы)
порок развития первого и второго

Синдром Ди Джорджи
Аплазия(полный) или
гипоплазия (частичный) тимуса
Триада -недоразвитие тимуса
-отсутствие
Синдром Ди Джорджи
Аплазия(полный) или
гипоплазия (частичный) тимуса
Триада -недоразвитие тимуса
-отсутствие
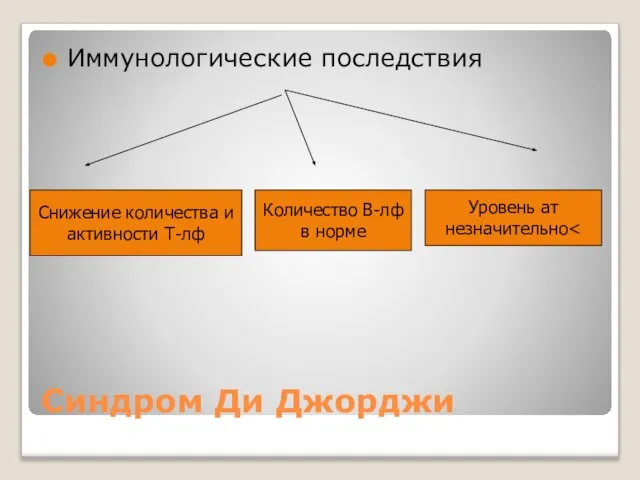
Синдром Ди Джорджи
Иммунологические последствия
Снижение количества и
активности Т-лф
Количество В-лф
в норме
Уровень
Синдром Ди Джорджи
Иммунологические последствия
Снижение количества и
активности Т-лф
Количество В-лф
в норме
Уровень

Синдром
Ди Джорджи
Клиника:
Рецидивирующие вирусные, паразитарные, вирусные инфекции
Судороги
Дисморфия лица
Другие пороки развития –атрезия
Синдром
Ди Джорджи
Клиника:
Рецидивирующие вирусные, паразитарные, вирусные инфекции
Судороги
Дисморфия лица
Другие пороки развития –атрезия

Больная с синдромом
Ди-Джорджи 11 лет
• ОРВИ 4-5 раз в год
• 10
Больная с синдромом
Ди-Джорджи 11 лет
• ОРВИ 4-5 раз в год
• 10

Синдром Незелофа
(лимфоцитарная дисгенезия, нормоплазмоцитарная и нормогаммаглобулинемическая аплазия)
Гипоплазия тимуса
АТ
Синдром Незелофа
(лимфоцитарная дисгенезия, нормоплазмоцитарная и нормогаммаглобулинемическая аплазия)
Гипоплазия тимуса
АТ
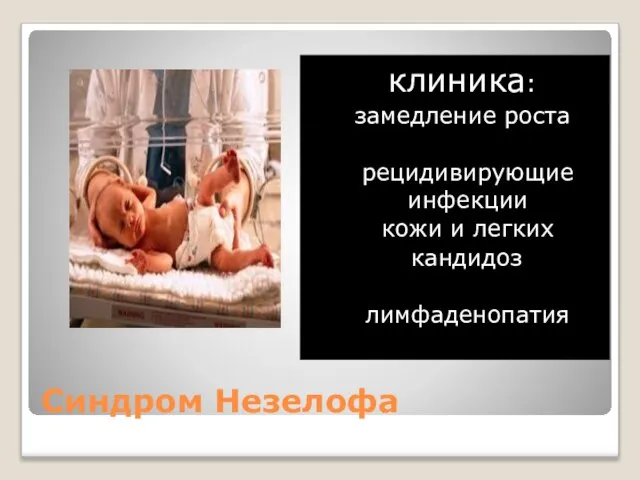
Синдром Незелофа
клиника:
замедление роста
рецидивирующие инфекции кожи и легких
кандидоз
Синдром Незелофа
клиника:
замедление роста
рецидивирующие инфекции кожи и легких
кандидоз

Дефицит В-клеточного звена
Агамма(гипогамма)глобулинемия (болезнь Брутона) рецессивный тип наследования
Дефицит цитоплазматической
тирозинкиназы
Нарушение дифференцировки
Дефицит В-клеточного звена
Агамма(гипогамма)глобулинемия (болезнь Брутона) рецессивный тип наследования
Дефицит цитоплазматической
тирозинкиназы
Нарушение дифференцировки

клиника
Определяется на 5-9 месяце жизни
Рецидивирующие гнойные инфекции
Менингиты
Отсутсвие реакции лф.узлов и селезенки
клиника
Определяется на 5-9 месяце жизни
Рецидивирующие гнойные инфекции
Менингиты
Отсутсвие реакции лф.узлов и селезенки

ТКИД – основные клинические проявления
• раннее начало (обычно между 3-9 месяцами
ТКИД – основные клинические проявления
• раннее начало (обычно между 3-9 месяцами

ТКИД (швейцарский тип)
Х-сцепленный тип
Дефект на уровне стволовой клетки
нежизнеспособность
ТКИД (швейцарский тип)
Х-сцепленный тип
Дефект на уровне стволовой клетки
нежизнеспособность

Синдром Вискотта-Олдрича
(СВО) - ТКИД
В основе лежит мутация гена,
который кодирует белок
Синдром Вискотта-Олдрича
(СВО) - ТКИД
В основе лежит мутация гена,
который кодирует белок

Комбинированные ИД
Синдром Вискотта-Олдрича
Нарушение активации Т-h и Т-s
<концентрация IgM, (IgA IgE >)
Комбинированные ИД
Синдром Вискотта-Олдрича
Нарушение активации Т-h и Т-s
<концентрация IgM, (IgA IgE >)

Синдром Вискотта-Олдрича
Клиника:
Экзема
Тромбоцитопения
Бактериальные и вирусные инфекции
Синдром Вискотта-Олдрича
Клиника:
Экзема
Тромбоцитопения
Бактериальные и вирусные инфекции

Больной с СВО 4-х лет
• С 3 месяцев до 3-х лет
Тяжелый
Больной с СВО 4-х лет
• С 3 месяцев до 3-х лет
Тяжелый
Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича

Синдром Луи-Барра
(атаксия-телеангиэктазия)
Гипоплазия тимуса
Гипоплазия лимф. узлов, селезенки
Количественная и функциональная недостаточность Т-лимфоцитов
Снижение количества
Синдром Луи-Барра
(атаксия-телеангиэктазия)
Гипоплазия тимуса
Гипоплазия лимф. узлов, селезенки
Количественная и функциональная недостаточность Т-лимфоцитов
Снижение количества

Синдром Луи-Барра
Клиника:
Атаксия
Телеангиэктазы
(на носу, ушах, конъюктиве)
Рецидивирующие инфекции
Опухолевые заболевания
Синдром Луи-Барра
Клиника:
Атаксия
Телеангиэктазы
(на носу, ушах, конъюктиве)
Рецидивирующие инфекции
Опухолевые заболевания
 Внешняя фиксация в травматологии
Внешняя фиксация в травматологии Холинергические средства
Холинергические средства Внутренняя среда. Кровь
Внутренняя среда. Кровь имобилизация и ее виды
имобилизация и ее виды Дисграфия
Дисграфия Аппараты, воспроизводящие движения нижней челюсти
Аппараты, воспроизводящие движения нижней челюсти Первая медицинская помощь при кровотечениях и ранениях
Первая медицинская помощь при кровотечениях и ранениях Пищеварительная система. Общие сведения
Пищеварительная система. Общие сведения Лекарственные формы с жидкой дисперсионной средой
Лекарственные формы с жидкой дисперсионной средой Оказание первой помощи при инородном теле верхних дыхательных путей
Оказание первой помощи при инородном теле верхних дыхательных путей Главная медицинская сестра стоматологии
Главная медицинская сестра стоматологии Deadliest diseases
Deadliest diseases Психопрофилактика болезней
Психопрофилактика болезней ReFace EyesLift. Мини-курс по омоложению области глаз
ReFace EyesLift. Мини-курс по омоложению области глаз Увеиты
Увеиты Ретинит, воспалительное заболевание сетчатки глаза
Ретинит, воспалительное заболевание сетчатки глаза Общие особенности инфекционных болезней
Общие особенности инфекционных болезней Тотальная внутривенная анестезия
Тотальная внутривенная анестезия Везикулярное заболевание свиней
Везикулярное заболевание свиней Igiena vieţii sexuale
Igiena vieţii sexuale Образец заполнения журналов по ведению дезинфекционного режима
Образец заполнения журналов по ведению дезинфекционного режима Ниволумаб в лечении местнораспространенного или метастатического рака легкого
Ниволумаб в лечении местнораспространенного или метастатического рака легкого Що ти вважаєш за цінність?
Що ти вважаєш за цінність? Первая медицинская помощь при травмах
Первая медицинская помощь при травмах Генитальды эндометриозы бар әйелдерді емдеуде
Генитальды эндометриозы бар әйелдерді емдеуде Сны и сновидения. 2 класс
Сны и сновидения. 2 класс Десмургия. Повязка. Перевязка
Десмургия. Повязка. Перевязка Бронхиальная астма
Бронхиальная астма