- Неэмпирическая квантовая химия

Содержание
- 2. молекулярная динамика и метод Монте-Карло неэмпирическая квантовая химия полуэмпирическая квантовая химия квантовая статистическая механика молекулярная механика
- 3. Неэмпирическая квантовая химия Исходные данные: координаты и сорт ядер молекулы, число электронов и базис, в котором
- 4. Орбитали гауссового типа (GTO или ОГТ) упрощают вычисление многоцентровых кулоновских и обменных интегралов. ОГТ отвечают потенциалу
- 5. Форма ОСT легко аппроксимируется суммой ОГT с различными экспонентами и коэффициентами. Поэтому в квантово-химических расчетах используются,
- 6. Первые базисные наборы были построены из ОГТ так, чтобы лучше всего описывать ОСТ. Сейчас базисные наборы
- 7. Терминология, используемая для базисов атомного типа Минимальный базисный набор – используется только одна функция на пару
- 8. Поляризационные и диффузные функции. При описании химической связи СОГТ, полученные из атомных ХФ расчетов, дополняют другими
- 9. Базисные наборы Попла - структура базисного набора задается для целой молекулы, а не для отдельных атомов
- 10. Некоторые рекомендуемые базисные наборы Попла для молекул, содержащих атомы с Z ≤ 9
- 11. Базисные наборы, рекомендуемые для описания свойств молекул
- 12. Деформационные электронные плотности молекулы СО (разности между электронной плотностью молекулы и плотностями составляющих ее атомов), вычисленные
- 13. Рассчитанные в различных базисах и экспериментальные характеристики молекулы СО Re – равновесное межъядерное расстояние, ωe –
- 14. Роль поляризационных функций при расчете инверсионного барьера нежесткой молекулы NH3 Потенциальные кривые инверсии молекулы аммиака. Расчет
- 16. Скачать презентацию
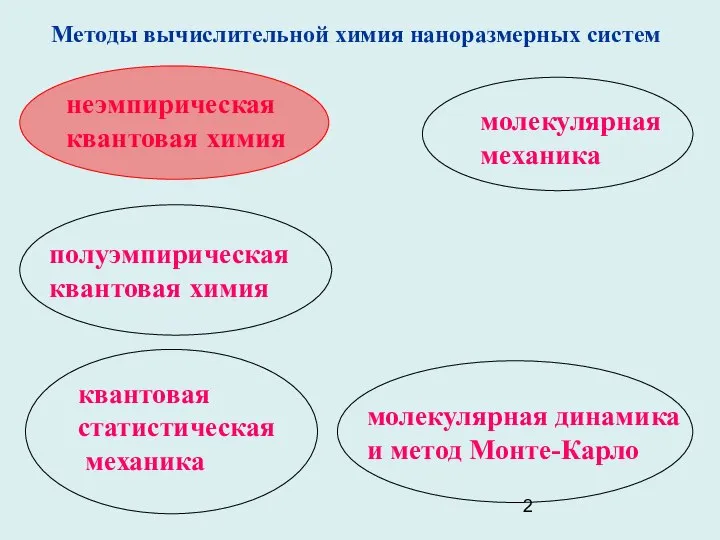
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия
молекулярная динамика
и метод Монте-Карло
неэмпирическая
квантовая химия
полуэмпирическая
квантовая химия

Неэмпирическая квантовая химия
Исходные данные: координаты и сорт ядер молекулы, число
Неэмпирическая квантовая химия
Исходные данные: координаты и сорт ядер молекулы, число
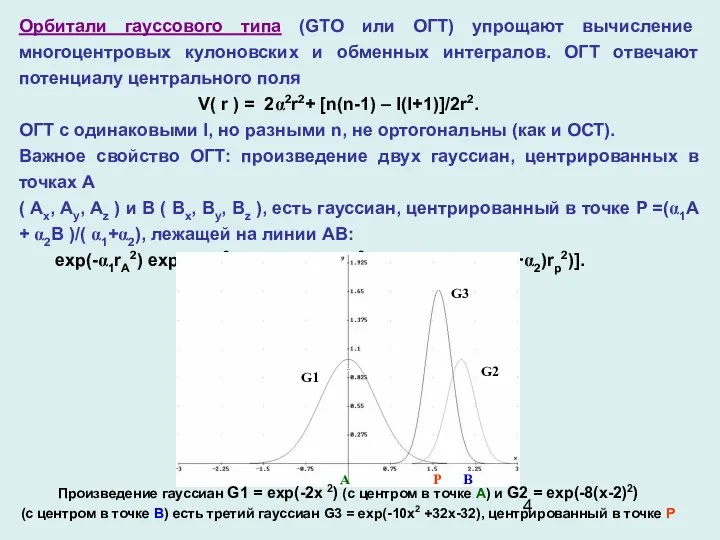
Орбитали гауссового типа (GTO или ОГТ) упрощают вычисление многоцентровых кулоновских и
Орбитали гауссового типа (GTO или ОГТ) упрощают вычисление многоцентровых кулоновских и

Форма ОСT легко аппроксимируется суммой ОГT с различными экспонентами и коэффициентами.
Форма ОСT легко аппроксимируется суммой ОГT с различными экспонентами и коэффициентами.

Первые базисные наборы были построены из ОГТ так, чтобы лучше всего
Первые базисные наборы были построены из ОГТ так, чтобы лучше всего

Терминология, используемая для базисов атомного типа
Минимальный базисный набор – используется только
Терминология, используемая для базисов атомного типа
Минимальный базисный набор – используется только

Поляризационные и диффузные функции. При описании химической связи СОГТ, полученные из
Поляризационные и диффузные функции. При описании химической связи СОГТ, полученные из

Базисные наборы Попла - структура базисного набора задается для целой молекулы,
Базисные наборы Попла - структура базисного набора задается для целой молекулы,
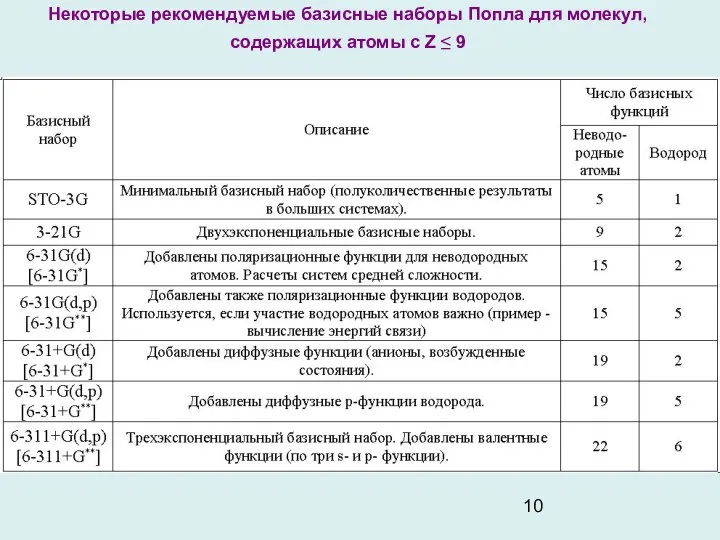
Некоторые рекомендуемые базисные наборы Попла для молекул, содержащих атомы с Z
Некоторые рекомендуемые базисные наборы Попла для молекул, содержащих атомы с Z
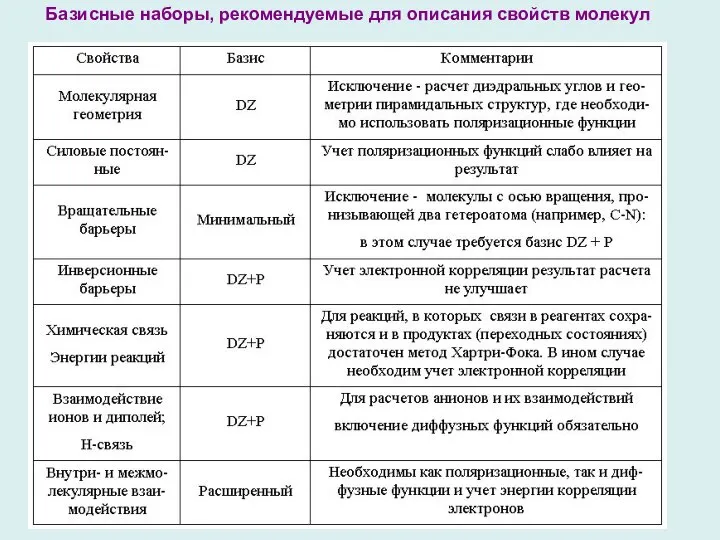
Базисные наборы, рекомендуемые для описания свойств молекул
Базисные наборы, рекомендуемые для описания свойств молекул
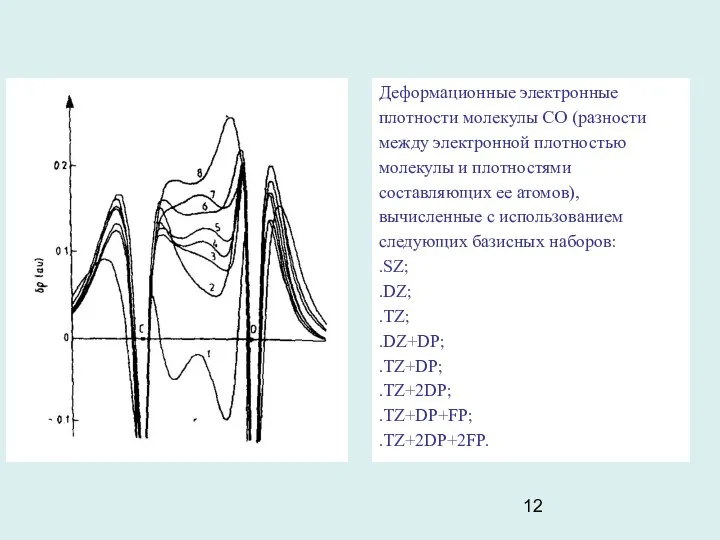
Деформационные электронные плотности молекулы СО (разности между электронной плотностью молекулы и
Деформационные электронные плотности молекулы СО (разности между электронной плотностью молекулы и
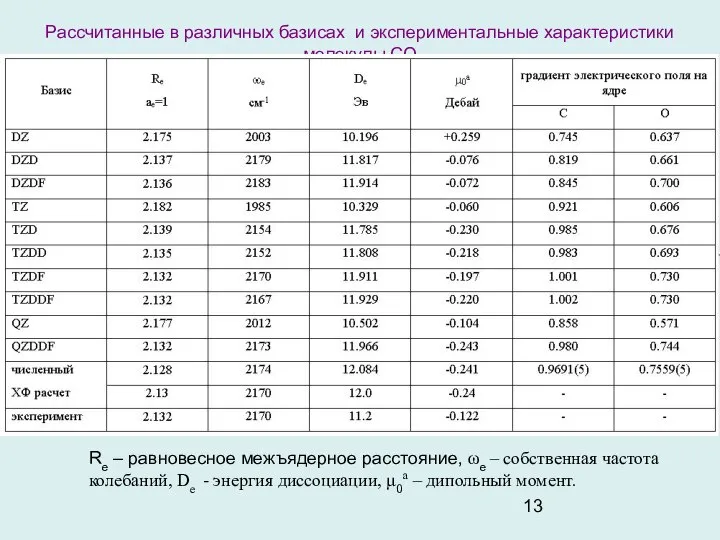
Рассчитанные в различных базисах и экспериментальные характеристики молекулы СО
Re – равновесное
Рассчитанные в различных базисах и экспериментальные характеристики молекулы СО
Re – равновесное
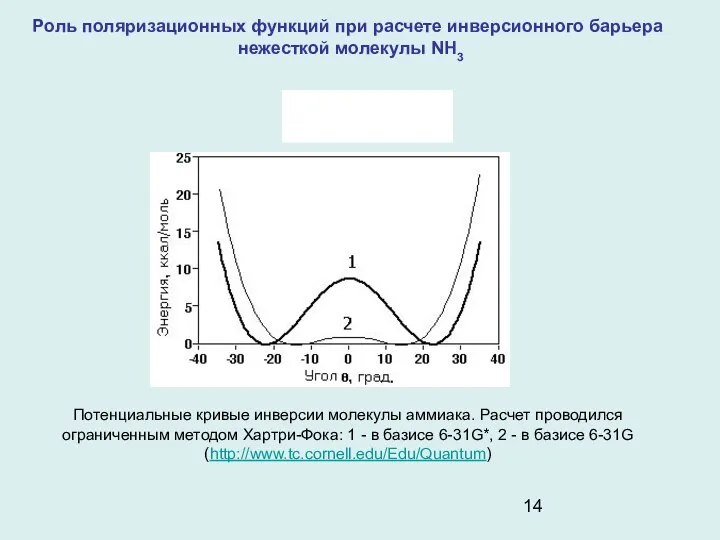
Роль поляризационных функций при расчете инверсионного барьера
нежесткой молекулы NH3
Потенциальные кривые
Роль поляризационных функций при расчете инверсионного барьера
нежесткой молекулы NH3
Потенциальные кривые
 Художественная обработка кожи
Художественная обработка кожи Флаги Тюмени и Тюменской области
Флаги Тюмени и Тюменской области Классный час : «Наша сила- в единстве» Подготовила учитель начальных классов МБОУ СОШ № 25 ст. Ладожской М.А.Ярушина
Классный час : «Наша сила- в единстве» Подготовила учитель начальных классов МБОУ СОШ № 25 ст. Ладожской М.А.Ярушина Презентация Классификация и кодирование товаров Лекция
Презентация Классификация и кодирование товаров Лекция Государственная служба в РФ
Государственная служба в РФ Лекарственное растительное сырье, содержащее эфирные масла
Лекарственное растительное сырье, содержащее эфирные масла Сталинград - презентация для начальной школы_
Сталинград - презентация для начальной школы_ Презентация на тему "Досвід роботи" - скачать презентации по Педагогике
Презентация на тему "Досвід роботи" - скачать презентации по Педагогике Непрерывная холодильная цепь
Непрерывная холодильная цепь Таможенный устав 1755 года Ростовцев А.Н. Т-102
Таможенный устав 1755 года Ростовцев А.Н. Т-102 Презентация на тему "Губский Л. В. - Современные методы нейровизуализации в диагностике ОНМК" - скачать презентации по Медицин
Презентация на тему "Губский Л. В. - Современные методы нейровизуализации в диагностике ОНМК" - скачать презентации по Медицин Проектная работа на тему Хоккей
Проектная работа на тему Хоккей  Міцність при змінних навантаженнях. (Лекція 3)
Міцність при змінних навантаженнях. (Лекція 3) Преодоление гравитации с помощью устройства ******
Преодоление гравитации с помощью устройства ****** Ключевые показатели работы КЦ
Ключевые показатели работы КЦ первая русская революция
первая русская революция Қазақстанның геосаяси жағдайын талдау
Қазақстанның геосаяси жағдайын талдау Гирдромеханические передачи
Гирдромеханические передачи Виды опросов
Виды опросов  История баскетбола
История баскетбола Семейное право
Семейное право Монополия в рыночной экономике
Монополия в рыночной экономике Философия Нового Времени XVI – XVIII вв.
Философия Нового Времени XVI – XVIII вв. 3 класс ─ маршрут-справочник
3 класс ─ маршрут-справочник Организм человека как единая саморазвивающаяся биологическая система (Лекция 4)
Организм человека как единая саморазвивающаяся биологическая система (Лекция 4) PR-кампании аналитический этап
PR-кампании аналитический этап Презентация Федеральный закон "О международных договорах Российской Федерации" от 15.07.1995 N 101-ФЗ
Презентация Федеральный закон "О международных договорах Российской Федерации" от 15.07.1995 N 101-ФЗ  Lingua araba 1 (Lingua e fonologia). Wael Farouq
Lingua araba 1 (Lingua e fonologia). Wael Farouq