- ЛИПИДОЗЫ

Содержание
- 2. Липидозы (lipidoses) — группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер. Большинство Л.
- 3. К липидозам относятся: ганглиозидозы — GM1-ганглиозидоз (тип I, II, III), GM2-ганглиозидоз (тип I, II, ювенильный, хронический
- 4. Ганглиозидозы — группа заболеваний, обусловленных нарушением катаболизма ганглиозидов — сложных гликолипидов, углеводная часть молекулы которых содержит
- 5. GM2-ганглиозидозы — большая фенотипически вариабельная группа заболеваний, в основе которых лежит дефицит гексозаминидазы А и В.
- 6. GM1-ганглиозидоз тип I (генерализованный семейный ганглиозидоз, нейровисцеральный липидоз, недостаточность b-галактозидазы) характеризуется аутосомно-рецессивно наследуемой недостаточностью фермента GM1-b-галактозадазы.
- 7. Клиника GM1-ганглиозидоз обнаруживается при рождении или вскоре после него. Отмечаются плохой аппетит, слабость сосания и крика,
- 8. Диагностика Диагноз GM1-ганглиозидоза подтверждается при определении активности b-галактозидазы в лейкоцитах и культивируемых фибробластах. GM1-ганглиозидоз тип I
- 9. Лечение: Специфическая терапия отсутствует. Разрабатываются методы заместительной терапии b-галактозидазой (очищенной или инкапсулированной в липосомах). Исход: Дети
- 10. GM1 ганглиозидоз тип II (ювенильный системный липидоз, болезнь Дерри) фенотипически отличается от GM1-ганглиозидоза тип I обычно
- 11. Клиника: В течение первого года жизни ребенок развивается нормально. К концу первого года возникают атаксия и
- 12. GM1-ганглиозидоз тип III проявляется прогрессирующей дизартрией на втором десятилетии жизни. Развиваются атаксия и спастические параличи, деменция,
- 13. GM2-ганглиозидоз тип I (амавротическая идиотия Тея — Сакса, болезнь Тея — Сакса) встречается преимущественно среди евреев-ашкенази,
- 14. В течение первых 4—6 месяцев жизни ребенок развивается нормально, начинает сидеть, ползать, улыбаться. Отмечается повышенная реакция
- 15. GM2-ганглиозидоз тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа) по клинической и патологоанатомической картине идентичен болезни Тея
- 16. Ювенильный GM2-ганглиозидоз. В основе его развития лежит дефицит фермента гексозаминидазы А. Этническая избирательность отсутствует. Первые симптомы
- 17. GM2-ганглиозидоз, хронический тип проявляется в возрасте от 4 до 16 лет медленно прогрессирующим нарушением походки. В
- 18. Болезнь Ниммана — Пика Впервые описана в 1914 г. немецким педиатром Ниманном (A. Niemann), а в
- 19. Болезнь Ниммана — Пика
- 20. Тип А (классическая инфантильная форма, острая нейропатическая форма) наблюдается наиболее часто. Заболевание проявляется после рождения и
- 21. При типе В (висцеральная форма, хроническая форма без вовлечения нервной системы) основные клинические проявления развиваются позже,
- 22. Болезнь Гоше глюкоцереброзидный липидоз, глюкоцереброзидоз — одно из наиболее частых наследственных нарушений гликолипидного обмена. Предполагают, что
- 23. Болезнь Гоше
- 24. Тип I (хронический, или взрослый, нейропатический) характеризуется развитием спленомегалии вскоре после рождения. В ряде случаев возможны
- 25. Тип II (острый инфантильный, острый нейропатический) может развиваться сразу после рождения и до 18 месяцев (в
- 26. Болезнь Фабри диффузная универсальная ангиокератома, наследственный дистонический липидоз — врожденный дефект катаболизма гликосфинголипидов с преобладанием поражения
- 27. Болезнь Фабри
- 28. У гомозиготных мужчин заболевание проявляется в детском возрасте или у подростков так называемыми кризами Фабри, характеризующимися
- 29. С возрастом увеличивается накопление сфингогликолипидов и гликопротеида в органах сердечно-сосудистой системы и почках. Появляются симптомы стенокардии,
- 30. Болезнь Краббе (глобоидно-клеточная лейкодистрофия) — быстро прогрессирующее демиелинизирующее дегенеративное заболевание ц.н.с. Тип наследования — аутосомно-рецессивный. В
- 32. Скачать презентацию

Липидозы (lipidoses) — группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно

К липидозам относятся:
ганглиозидозы — GM1-ганглиозидоз (тип I, II, III), GM2-ганглиозидоз (тип
К липидозам относятся:
ганглиозидозы — GM1-ганглиозидоз (тип I, II, III), GM2-ганглиозидоз (тип

Ганглиозидозы — группа заболеваний, обусловленных нарушением катаболизма ганглиозидов — сложных гликолипидов, углеводная часть
Ганглиозидозы — группа заболеваний, обусловленных нарушением катаболизма ганглиозидов — сложных гликолипидов, углеводная часть

GM2-ганглиозидозы — большая фенотипически вариабельная группа заболеваний, в основе которых лежит дефицит
GM2-ганглиозидозы — большая фенотипически вариабельная группа заболеваний, в основе которых лежит дефицит

GM1-ганглиозидоз тип I (генерализованный семейный ганглиозидоз, нейровисцеральный липидоз, недостаточность b-галактозидазы) характеризуется
GM1-ганглиозидоз тип I (генерализованный семейный ганглиозидоз, нейровисцеральный липидоз, недостаточность b-галактозидазы) характеризуется

Клиника
GM1-ганглиозидоз обнаруживается при рождении или вскоре после него. Отмечаются плохой
Клиника
GM1-ганглиозидоз обнаруживается при рождении или вскоре после него. Отмечаются плохой

Диагностика
Диагноз GM1-ганглиозидоза подтверждается при определении активности b-галактозидазы в лейкоцитах и
Диагностика
Диагноз GM1-ганглиозидоза подтверждается при определении активности b-галактозидазы в лейкоцитах и

Лечение:
Специфическая терапия отсутствует. Разрабатываются методы заместительной терапии b-галактозидазой (очищенной или инкапсулированной
Лечение:
Специфическая терапия отсутствует. Разрабатываются методы заместительной терапии b-галактозидазой (очищенной или инкапсулированной

GM1 ганглиозидоз тип II (ювенильный системный липидоз, болезнь Дерри) фенотипически отличается от
GM1 ганглиозидоз тип II (ювенильный системный липидоз, болезнь Дерри) фенотипически отличается от

Клиника:
В течение первого года жизни ребенок развивается нормально. К концу
Клиника:
В течение первого года жизни ребенок развивается нормально. К концу

GM1-ганглиозидоз тип III проявляется прогрессирующей дизартрией на втором десятилетии жизни. Развиваются
GM1-ганглиозидоз тип III проявляется прогрессирующей дизартрией на втором десятилетии жизни. Развиваются

GM2-ганглиозидоз тип I (амавротическая идиотия Тея — Сакса, болезнь Тея — Сакса) встречается
GM2-ганглиозидоз тип I (амавротическая идиотия Тея — Сакса, болезнь Тея — Сакса) встречается

В течение первых 4—6 месяцев жизни ребенок развивается нормально, начинает сидеть, ползать,
В течение первых 4—6 месяцев жизни ребенок развивается нормально, начинает сидеть, ползать,

GM2-ганглиозидоз тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа) по клинической и
GM2-ганглиозидоз тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа) по клинической и

Ювенильный GM2-ганглиозидоз. В основе его развития лежит дефицит фермента гексозаминидазы А.
Ювенильный GM2-ганглиозидоз. В основе его развития лежит дефицит фермента гексозаминидазы А.

GM2-ганглиозидоз, хронический тип проявляется в возрасте от 4 до 16 лет медленно прогрессирующим
GM2-ганглиозидоз, хронический тип проявляется в возрасте от 4 до 16 лет медленно прогрессирующим

Болезнь Ниммана — Пика
Впервые описана в 1914 г. немецким педиатром Ниманном
Болезнь Ниммана — Пика
Впервые описана в 1914 г. немецким педиатром Ниманном

Болезнь Ниммана — Пика
Болезнь Ниммана — Пика

Тип А (классическая инфантильная форма, острая нейропатическая форма) наблюдается наиболее часто.
Тип А (классическая инфантильная форма, острая нейропатическая форма) наблюдается наиболее часто.

При типе В (висцеральная форма, хроническая форма без вовлечения нервной системы)
При типе В (висцеральная форма, хроническая форма без вовлечения нервной системы)

Болезнь Гоше
глюкоцереброзидный липидоз, глюкоцереброзидоз — одно из наиболее частых наследственных нарушений гликолипидного
Болезнь Гоше
глюкоцереброзидный липидоз, глюкоцереброзидоз — одно из наиболее частых наследственных нарушений гликолипидного
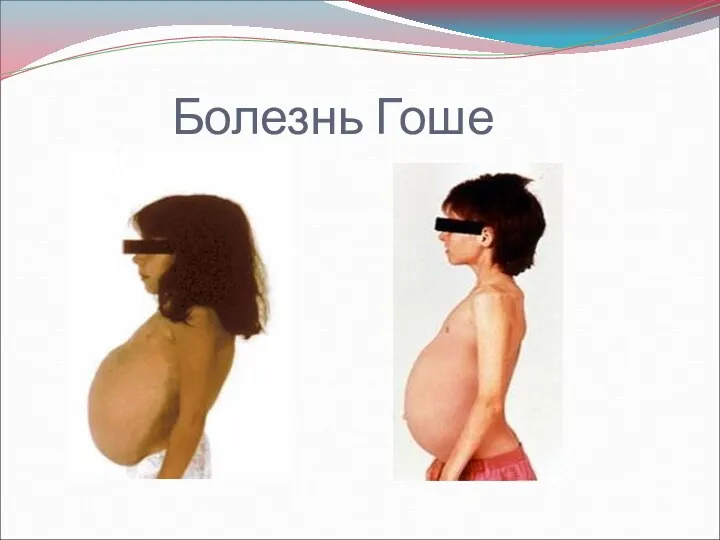
Болезнь Гоше
Болезнь Гоше

Тип I (хронический, или взрослый, нейропатический) характеризуется развитием спленомегалии вскоре после
Тип I (хронический, или взрослый, нейропатический) характеризуется развитием спленомегалии вскоре после

Тип II (острый инфантильный, острый нейропатический) может развиваться сразу после рождения
Тип II (острый инфантильный, острый нейропатический) может развиваться сразу после рождения

Болезнь Фабри
диффузная универсальная ангиокератома, наследственный дистонический липидоз — врожденный дефект катаболизма гликосфинголипидов
Болезнь Фабри
диффузная универсальная ангиокератома, наследственный дистонический липидоз — врожденный дефект катаболизма гликосфинголипидов

Болезнь Фабри
Болезнь Фабри

У гомозиготных мужчин заболевание проявляется в детском возрасте или у подростков
У гомозиготных мужчин заболевание проявляется в детском возрасте или у подростков

С возрастом увеличивается накопление сфингогликолипидов и гликопротеида в органах сердечно-сосудистой системы
С возрастом увеличивается накопление сфингогликолипидов и гликопротеида в органах сердечно-сосудистой системы

Болезнь Краббе (глобоидно-клеточная лейкодистрофия) — быстро прогрессирующее демиелинизирующее дегенеративное заболевание ц.н.с. Тип
Болезнь Краббе (глобоидно-клеточная лейкодистрофия) — быстро прогрессирующее демиелинизирующее дегенеративное заболевание ц.н.с. Тип
 Отличительные символы России. Народы России. Загадки
Отличительные символы России. Народы России. Загадки Разработка робота на основе платформы Аrduino в качестве учебного пособия для МДК 02.03 «Мехатроника и робототехника»
Разработка робота на основе платформы Аrduino в качестве учебного пособия для МДК 02.03 «Мехатроника и робототехника» Австрийская (психологическая) школа маржинализма Выполнили: Студенты 1 курса Экономического факультета Группы М 122 Б Зелепукин
Австрийская (психологическая) школа маржинализма Выполнили: Студенты 1 курса Экономического факультета Группы М 122 Б Зелепукин  «Простота» тестирования небольшого системного ПО Калугин Александр, PhD, PMP Mercury Development Project Director
«Простота» тестирования небольшого системного ПО Калугин Александр, PhD, PMP Mercury Development Project Director Не расточая лишних слов, Я вам дарю букет цветов. Желаю быть прекрасным дамам Ещё прекраснее - с цветами! Для Вас сегодня солнце свет
Не расточая лишних слов, Я вам дарю букет цветов. Желаю быть прекрасным дамам Ещё прекраснее - с цветами! Для Вас сегодня солнце свет Болезни органов дыхания и их предупреждение Подготовила Ученица 8 класса «А» МОУ «Гимназия №4» Гоова Назима
Болезни органов дыхания и их предупреждение Подготовила Ученица 8 класса «А» МОУ «Гимназия №4» Гоова Назима Преобразование пешеходной ул. Ленина в г. Горнозаводск
Преобразование пешеходной ул. Ленина в г. Горнозаводск Имя гражданина. Право на имя. _
Имя гражданина. Право на имя. _ «Экономика организации» На тему: Организация производства. Типы производственных структур. Выполнила: студентка
«Экономика организации» На тему: Организация производства. Типы производственных структур. Выполнила: студентка  Презентация на тему "Лекция 17. Методические и технические аспекты холтеровского мониторирования ЭКГ" - скачать презентации п
Презентация на тему "Лекция 17. Методические и технические аспекты холтеровского мониторирования ЭКГ" - скачать презентации п Древнерусское искусство. Храмовая архитектура
Древнерусское искусство. Храмовая архитектура Графический учебный исполнитель
Графический учебный исполнитель Развитие творческих способностей младших школьников
Развитие творческих способностей младших школьников 1нче бүлек. Фәлсәфәгә кереш
1нче бүлек. Фәлсәфәгә кереш Массивы - операции с массивами (размер изменяемый)
Массивы - операции с массивами (размер изменяемый) Резьбы. Резьбовые изделия и соединения
Резьбы. Резьбовые изделия и соединения Лекция 8 Смазки
Лекция 8 Смазки Имущественное страхование
Имущественное страхование Презентация Дебиторская задолженность
Презентация Дебиторская задолженность  Модель диагностики изменений Надлера и Ташмена Подготовили: студентки группы ДС04, Шунайлова Жанна и Епифанова Евгения
Модель диагностики изменений Надлера и Ташмена Подготовили: студентки группы ДС04, Шунайлова Жанна и Епифанова Евгения Охрана атмосферного воздуха в городах и других населенных пунктах Выполнила студентка группы Ю124Б Злобарь Софья
Охрана атмосферного воздуха в городах и других населенных пунктах Выполнила студентка группы Ю124Б Злобарь Софья Обеззараживание воды
Обеззараживание воды Вид спорта бобслей ,
Вид спорта бобслей , Алгебраические уравнения произвольных степеней
Алгебраические уравнения произвольных степеней Организация АС и ДНР при аварии в городе
Организация АС и ДНР при аварии в городе Технология возведения заглубленных сооружений
Технология возведения заглубленных сооружений Slozhnye_polufabrikaty
Slozhnye_polufabrikaty 2010/2011 учебный год
2010/2011 учебный год