- La reazione a catena della DNA polimerasi

Содержание
- 2. PCR “polymerase chain reaction” Descrizione della tecnica, metodo, componenti, variabili, strumenti = termociclatori Tecnica: amplificazione esponenziale
- 3. Come si fa la PCR, in cosa consiste amplificazione tramite sintesi de-novo di nuovi filamenti di
- 4. 1 denaturazione del DNA a 94°C 2 appaiamento (“annealing”) dei primers al ssDNA templato, temperatura misurata
- 5. applicazione del sistema naturale la sintesi del DNA è semiconservativa (duplicazione) le DNA polimerasi e anche
- 6. l’invenzione geniale dalla amplificazione lineare a quella esponenziale: la sintesi in vitro di DNA già era
- 7. attenzione al verso dei primers la doppia elica di DNA ha i due filamenti antiparalleli dovuti
- 8. se la reazione in vitro continua 1 doppia elica 2 doppie eliche
- 9. diventa una reazione esponenziale ma come è possibile fare avvenire la reazione senza farla fermare ?
- 10. 5’ 5’ 5’ 3’ 3’ 3’ 3’ 3’ 3’ 5’ 5’ 5’ Denaturazione Extension (Polimerizzazione) Annealing
- 11. Annealing Extension (Polimerizzazione) La crescita e’ esponenziale : teoricamente ad ogni ciclo un raddoppio di DNA,
- 12. vanno determinate per i tempi, temperature e quantita’(conc.) ogni ciclo = 3 passaggi (fasi) I passaggio
- 13. l’enzima sarà una Taq polimerasi (ne esistono diverse ottenute da mutanti che aumentano la specificità, efficienza
- 14. Volume di reazione: da 10 a 50 μl (max 100 μl) per una preparativa. Il DNA
- 15. la PCR si utilizza per amplificare i frammenti random da sequenziare a cui sono stati attaccati
- 16. applicazioni della PCR RT-PCR, nested PCR PCR inversa RACE 3’ e RACE 5’ variazioni sul tema
- 17. Ricerca di un vettore Per inserzione random nel genoma Per inserzione sito specifica nel genoma Cosa
- 18. Cambia la regione limitrofa Gene targeting deve avere regioni limitrofe note a) Random recombination ha regioni
- 19. Altre possibilità di analisi tramite PCR perfezionamenti delle tecniche e degli enzimi - nuove macchine con
- 20. Altre possibili applicazioni della PCR - abbiamo visto RT-PCR tramite reverse transcriptase da mRNA RT-PCR è
- 21. applicazione RT-PCR nuovo esercizio: se devo retrotrascrivere un mRNA per ottenere un un cDNA devo fare
- 22. La reverse trascrittasi RT L’uso della reverse trascrittasi risale a quando furono scoperti i meccanismi molecolari
- 23. RT-PCR: cosa si analizza = analisi della trascrizione di un gene o isolamento di un cDNA
- 24. stratagemmi della RT-PCR Accorgimento: quando si estrae l’RNA si deve evitare il DNA e si puo’
- 25. la retrotrascrizione Per RT si intende reverse transcriptase su templato di RNA Per avere un cDNA
- 26. vantaggi della RT-PCR Analisi della trascrizione tramite PCR Analisi della trascrizione e non determinazione del PM
- 27. come si fa una RT-PCR Si deve ottenere il retrotrascritto cioè il cDNA ( DNA complementare
- 28. RT-PCR dal II filamento in poi Accorgimenti e controlli della RT-PCR Prima di retrotrascrivere il cDNA
- 29. genomic & coding sequence, mRNA, cDNA 5’ 3’ 5’ 3’ PCR su cDNA a due filamenti
- 30. Correzione parametri di una PCR La PCR deve dare dei prodotti che corrispondono agli attesi Quando
- 31. sequenza di un cDNA dalla banca dati per aumentare specificità? facciamo una doppia PCR : la
- 32. la sequenza 5’- 3’ di un cDNA GGATCCCTGT TCCTGATCAC TGATCTCTGG TTCTTTTATT ATGCATATTC 50 ATTTTGAAAT CTGATTCCTT TTCTGAGCAT
- 33. una nested PCR 1 gatcacaggt ctatcaccct attaaccact cacgggagct ctccatgcat ttggtatttt 61 cgtctggggg gtgtgcacgc gatagcattg cgagacgctg gagccggagc
- 34. i controlli essenziali Controlli, negativi, positivi, (i controlli ci fanno capire se l’esperimento è venuto bene)
- 35. procedure la Rev Transcript virale a 37°C, mutanti max 65°C oligo dT, random priming con esanucleotidi,
- 36. precauzioni estrarre RNA eliminando DNA genomico che falsifica il risultato cosa si vuole vedere con RT-PCR:
- 37. può essere quantitativa? la RT-PCR può essere quantitativa l’amplificazione è proporzionale alla quantità di templato l’amplificazione
- 38. perchè quantitativa ? l’amplificazione è proporzionale al templato iniziale, perchè si conserva la proporzionalità anche dopo
- 39. la rivelazione su gel dopo elettroforesi su gel di agarosio si rivela il DNA amplificato come
- 40. controllo di RT-PCR altro controllo negativo : assenza di amplificazione sui campioni di RNA non retrotrascritti
- 41. Controlli della PCR Controlli di estrazione: quali? ripetibilità della amplificazione ripetibilità su campioni indipendenti univocità di
- 42. R.A.C.E. Con la RT-PCR si amplifica solo un frammento del cDNA Se si vuole identificare l’intero
- 43. (Rapid Amplification of cDNA Ends) La RACE è una tecnica per l’amplificazione di sequenze nucleotidiche, usando
- 44. Differenze nella ricerca di 5’ o 3’ ignoti mRNA AAAAA 5’ 3’ poly TTTTT 5’ 3’
- 45. Cerchiamo il 3’ sconosciuto In questo caso si usa un oligo dT per sintetizzare il cDNA
- 46. Dalle banche EST (expressed sequence tags); Da studi di funzione (per esempio EXON e PROMOTER TRAPPING);
- 47. mRNA poly A 5’ noto 3’ ignoto sintesi del I filamento di cDNA con RT con
- 48. Scelta dei primers per la RACE 3’ Il primo primer obbligato è quello per la RT
- 49. RACE 3’ TTT…..TTT 5’ 5’ mRNA poly(A) tail 1 - Annealing tra la coda di polyA
- 50. Nested PCR o PCR interna cDNA 5’ 3’ TTTTT regione nota regione ignota I primer specifico
- 51. un trucco che inganna la polimerasi 3’ 5’ 5’ 3’ primer la polimerase estende da un
- 52. 2 - Degradazione del templato di RNA 3 - Amplificazione per PCR usando un primer specifico
- 53. GSP 2 TTT…..TTT 5’ 3’ AUAP UAP 3’ 5’ La SPECIFICITA’ può essere garantita da un
- 54. PCR nested RACE 3’ Nel caso di una RACE 3’ cambia solo il verso dei primers
- 55. RACE 5’ Cerchiamo il 5’ ignoto Dobbiamo comunque ottenere il cDNA ed utiliziamo gli stessi metodi
- 56. 5’ mRNA poly(A) tail 1 - Annealing tra una regione interna dell’mRNA e un primer gene
- 57. 2 - Retrotrascrizione e tailing all’estremità 3’ del cDNA 5’ AAA….AAA n 5’ 3’ Degradazione mRNA
- 58. Race 5’ fase III I inosina
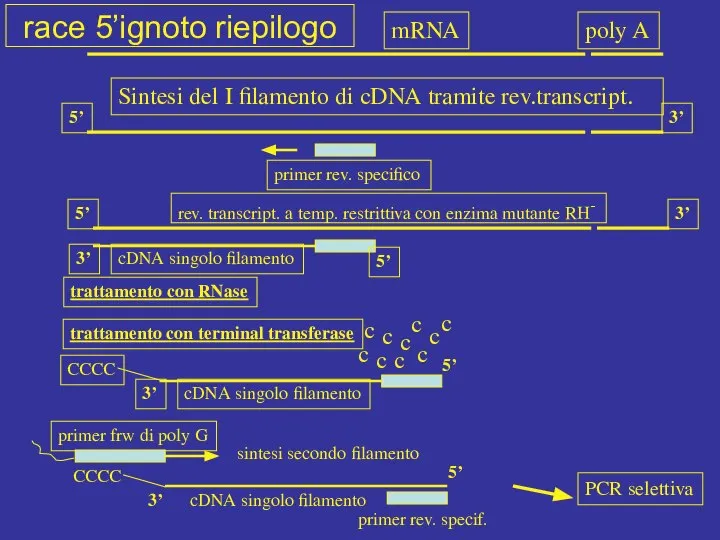
- 59. mRNA poly A Sintesi del I filamento di cDNA tramite rev.transcript. primer rev. specifico 5’ 3’
- 60. dopo la sintesi del I filam. di cDNA e la terminal transferase, PCR selettiva con II
- 62. Скачать презентацию

PCR “polymerase chain reaction”
Descrizione della tecnica, metodo, componenti, variabili, strumenti =
PCR “polymerase chain reaction”
Descrizione della tecnica, metodo, componenti, variabili, strumenti =

Come si fa la PCR, in cosa consiste
amplificazione tramite sintesi
Come si fa la PCR, in cosa consiste
amplificazione tramite sintesi

1 denaturazione del DNA a 94°C
2 appaiamento (“annealing”) dei primers al
1 denaturazione del DNA a 94°C
2 appaiamento (“annealing”) dei primers al

applicazione del sistema naturale
la sintesi del DNA è semiconservativa (duplicazione)
le
applicazione del sistema naturale
la sintesi del DNA è semiconservativa (duplicazione)
le

l’invenzione geniale
dalla amplificazione lineare a quella esponenziale:
la sintesi in vitro di
l’invenzione geniale
dalla amplificazione lineare a quella esponenziale:
la sintesi in vitro di
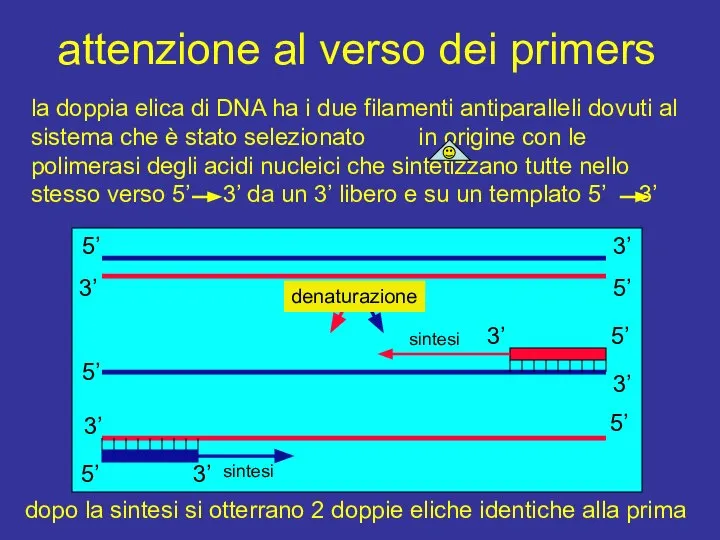
attenzione al verso dei primers
la doppia elica di DNA ha i
attenzione al verso dei primers
la doppia elica di DNA ha i
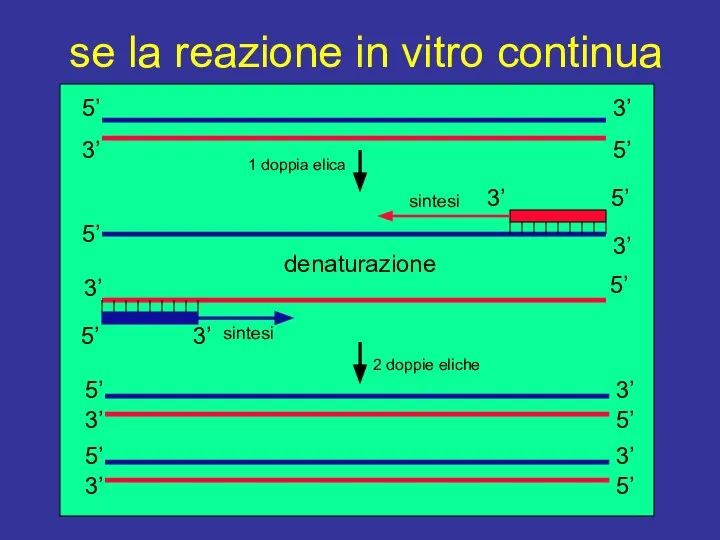
se la reazione in vitro continua
1 doppia elica
2 doppie eliche
se la reazione in vitro continua
1 doppia elica
2 doppie eliche
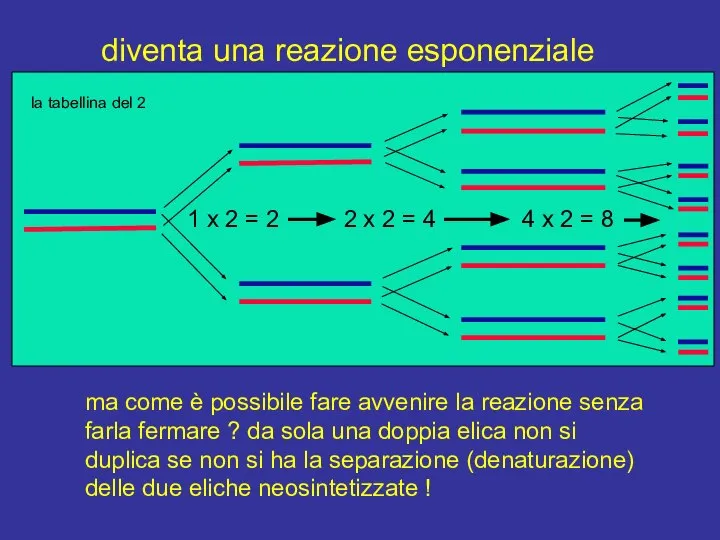
diventa una reazione esponenziale
ma come è possibile fare avvenire la
diventa una reazione esponenziale
ma come è possibile fare avvenire la
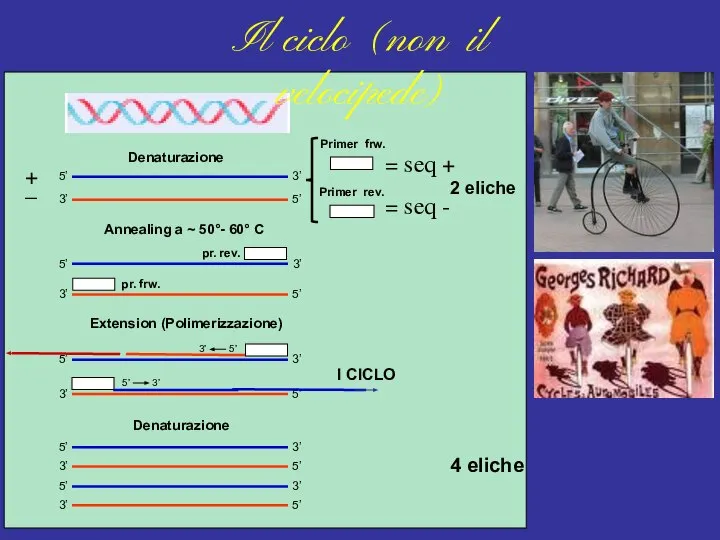
5’
5’
5’
3’
3’
3’
3’
3’
3’
5’
5’
5’
Denaturazione
Extension (Polimerizzazione)
Annealing a ~ 50°- 60° C
Denaturazione
5’
5’
5’
3’
3’
3’
3’
5’
Primer frw.
Primer rev.
I CICLO
2 eliche
4
5’
5’
5’
3’
3’
3’
3’
3’
3’
5’
5’
5’
Denaturazione
Extension (Polimerizzazione)
Annealing a ~ 50°- 60° C
Denaturazione
5’
5’
5’
3’
3’
3’
3’
5’
Primer frw.
Primer rev.
I CICLO
2 eliche
4
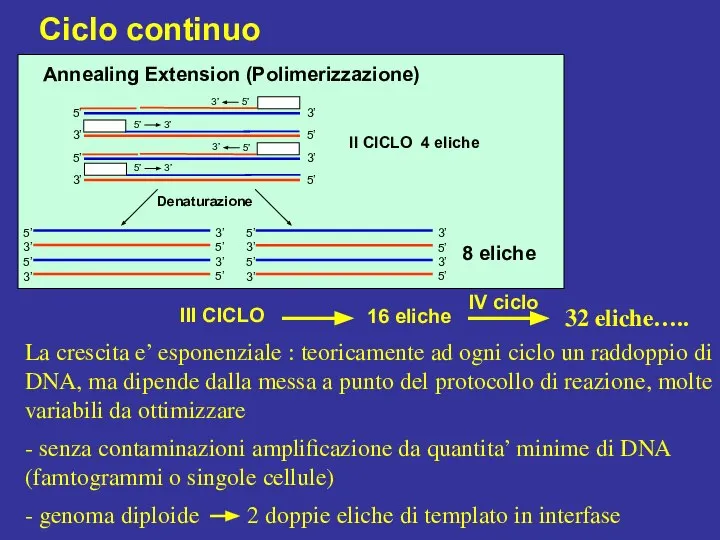
Annealing Extension (Polimerizzazione)
La crescita e’ esponenziale : teoricamente ad ogni ciclo
Annealing Extension (Polimerizzazione)
La crescita e’ esponenziale : teoricamente ad ogni ciclo

vanno determinate per i tempi, temperature e quantita’(conc.) ogni ciclo =
vanno determinate per i tempi, temperature e quantita’(conc.) ogni ciclo =

l’enzima sarà una Taq polimerasi (ne esistono diverse ottenute da mutanti
l’enzima sarà una Taq polimerasi (ne esistono diverse ottenute da mutanti

Volume di reazione: da 10 a 50 μl (max 100 μl)
Volume di reazione: da 10 a 50 μl (max 100 μl)

la PCR si utilizza per amplificare i frammenti random da sequenziare
la PCR si utilizza per amplificare i frammenti random da sequenziare

applicazioni della PCR
RT-PCR, nested PCR
PCR inversa
RACE 3’ e RACE 5’
variazioni
applicazioni della PCR
RT-PCR, nested PCR
PCR inversa
RACE 3’ e RACE 5’
variazioni

Ricerca di un vettore
Per inserzione random nel genoma
Per inserzione sito specifica
Ricerca di un vettore
Per inserzione random nel genoma
Per inserzione sito specifica

Cambia la regione limitrofa
Gene targeting deve avere regioni limitrofe note a)
Random
Cambia la regione limitrofa
Gene targeting deve avere regioni limitrofe note a)
Random

Altre possibilità di analisi tramite PCR
perfezionamenti delle tecniche e
Altre possibilità di analisi tramite PCR
perfezionamenti delle tecniche e

Altre possibili applicazioni della PCR
- abbiamo visto RT-PCR
tramite reverse transcriptase da
Altre possibili applicazioni della PCR
- abbiamo visto RT-PCR
tramite reverse transcriptase da

applicazione RT-PCR
nuovo esercizio: se devo retrotrascrivere un mRNA per ottenere un
applicazione RT-PCR
nuovo esercizio: se devo retrotrascrivere un mRNA per ottenere un

La reverse trascrittasi RT
L’uso della reverse trascrittasi risale a quando furono
La reverse trascrittasi RT
L’uso della reverse trascrittasi risale a quando furono

RT-PCR: cosa si analizza
= analisi della trascrizione di un gene
RT-PCR: cosa si analizza
= analisi della trascrizione di un gene

stratagemmi della RT-PCR
Accorgimento: quando si estrae l’RNA si deve evitare il
stratagemmi della RT-PCR
Accorgimento: quando si estrae l’RNA si deve evitare il

la retrotrascrizione
Per RT si intende reverse transcriptase su templato di RNA
la retrotrascrizione
Per RT si intende reverse transcriptase su templato di RNA

vantaggi della RT-PCR
Analisi della trascrizione tramite PCR
Analisi della trascrizione e non
vantaggi della RT-PCR
Analisi della trascrizione tramite PCR
Analisi della trascrizione e non

come si fa una RT-PCR
Si deve ottenere il retrotrascritto cioè il
come si fa una RT-PCR
Si deve ottenere il retrotrascritto cioè il

RT-PCR dal II filamento in poi
Accorgimenti e controlli della RT-PCR
Prima di
RT-PCR dal II filamento in poi
Accorgimenti e controlli della RT-PCR
Prima di
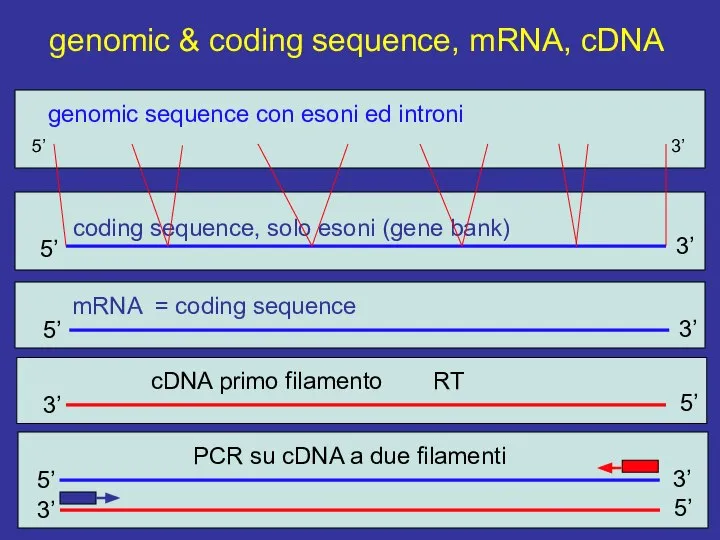
genomic & coding sequence, mRNA, cDNA
5’
3’
5’
3’
PCR su cDNA a due filamenti
genomic
genomic & coding sequence, mRNA, cDNA
5’
3’
5’
3’
PCR su cDNA a due filamenti
genomic

Correzione parametri di una PCR
La PCR deve dare dei prodotti che
Correzione parametri di una PCR
La PCR deve dare dei prodotti che

sequenza di un cDNA dalla banca dati
per aumentare specificità?
facciamo una doppia
sequenza di un cDNA dalla banca dati
per aumentare specificità?
facciamo una doppia

la sequenza 5’- 3’ di un cDNA
GGATCCCTGT TCCTGATCAC TGATCTCTGG TTCTTTTATT ATGCATATTC
la sequenza 5’- 3’ di un cDNA
GGATCCCTGT TCCTGATCAC TGATCTCTGG TTCTTTTATT ATGCATATTC

una nested PCR
1 gatcacaggt ctatcaccct attaaccact cacgggagct ctccatgcat ttggtatttt
61
una nested PCR
1 gatcacaggt ctatcaccct attaaccact cacgggagct ctccatgcat ttggtatttt
61

i controlli essenziali
Controlli, negativi, positivi,
(i controlli ci fanno capire se l’esperimento
i controlli essenziali
Controlli, negativi, positivi,
(i controlli ci fanno capire se l’esperimento

procedure
la Rev Transcript virale a 37°C, mutanti max 65°C
oligo dT, random
procedure
la Rev Transcript virale a 37°C, mutanti max 65°C
oligo dT, random

precauzioni
estrarre RNA eliminando DNA genomico che falsifica il risultato
cosa si vuole
precauzioni
estrarre RNA eliminando DNA genomico che falsifica il risultato
cosa si vuole

può essere quantitativa?
la RT-PCR può essere quantitativa
l’amplificazione è proporzionale alla quantità
può essere quantitativa?
la RT-PCR può essere quantitativa
l’amplificazione è proporzionale alla quantità

perchè quantitativa ?
l’amplificazione è proporzionale al templato iniziale,
perchè si conserva
perchè quantitativa ?
l’amplificazione è proporzionale al templato iniziale,
perchè si conserva

la rivelazione su gel
dopo elettroforesi su gel di agarosio si rivela
la rivelazione su gel
dopo elettroforesi su gel di agarosio si rivela

controllo di RT-PCR
altro controllo negativo :
assenza di amplificazione sui campioni di
controllo di RT-PCR
altro controllo negativo :
assenza di amplificazione sui campioni di

Controlli della PCR
Controlli di estrazione: quali?
ripetibilità della amplificazione
ripetibilità su
Controlli della PCR
Controlli di estrazione: quali?
ripetibilità della amplificazione
ripetibilità su

R.A.C.E.
Con la RT-PCR si amplifica solo un frammento del cDNA
Se si
R.A.C.E.
Con la RT-PCR si amplifica solo un frammento del cDNA
Se si

(Rapid Amplification of cDNA Ends)
La RACE è una tecnica per l’amplificazione
(Rapid Amplification of cDNA Ends)
La RACE è una tecnica per l’amplificazione

Differenze nella ricerca di 5’ o 3’ ignoti
mRNA
AAAAA
5’
3’
poly TTTTT
5’
3’
oligo esa nucleotidi
Differenze nella ricerca di 5’ o 3’ ignoti
mRNA
AAAAA
5’
3’
poly TTTTT
5’
3’
oligo esa nucleotidi

Cerchiamo il 3’ sconosciuto
In questo caso si usa un oligo dT
Cerchiamo il 3’ sconosciuto
In questo caso si usa un oligo dT

Dalle banche EST (expressed sequence tags);
Da studi di funzione (per
Dalle banche EST (expressed sequence tags);
Da studi di funzione (per

mRNA
poly A
5’ noto
3’ ignoto
sintesi del I filamento di cDNA con RT
mRNA
poly A
5’ noto
3’ ignoto
sintesi del I filamento di cDNA con RT

Scelta dei primers per la RACE 3’
Il primo primer obbligato
Scelta dei primers per la RACE 3’
Il primo primer obbligato

RACE 3’
TTT…..TTT
5’
5’
mRNA
poly(A) tail
1 - Annealing tra la coda di polyA
RACE 3’
TTT…..TTT
5’
5’
mRNA
poly(A) tail
1 - Annealing tra la coda di polyA

Nested PCR o PCR interna
cDNA
5’
3’
TTTTT
regione nota
regione ignota
I primer specifico
II primer specifico
Il
Nested PCR o PCR interna
cDNA
5’
3’
TTTTT
regione nota
regione ignota
I primer specifico
II primer specifico
Il

un trucco che inganna la polimerasi
3’
5’
5’
3’
primer
la polimerase estende da un 3’
un trucco che inganna la polimerasi
3’
5’
5’
3’
primer
la polimerase estende da un 3’

2 - Degradazione del templato di RNA
3 - Amplificazione per PCR
2 - Degradazione del templato di RNA
3 - Amplificazione per PCR

GSP 2
TTT…..TTT
5’
3’
AUAP
UAP
3’
5’
La SPECIFICITA’ può essere garantita da un ulteriore amplificazione con
GSP 2
TTT…..TTT
5’
3’
AUAP
UAP
3’
5’
La SPECIFICITA’ può essere garantita da un ulteriore amplificazione con
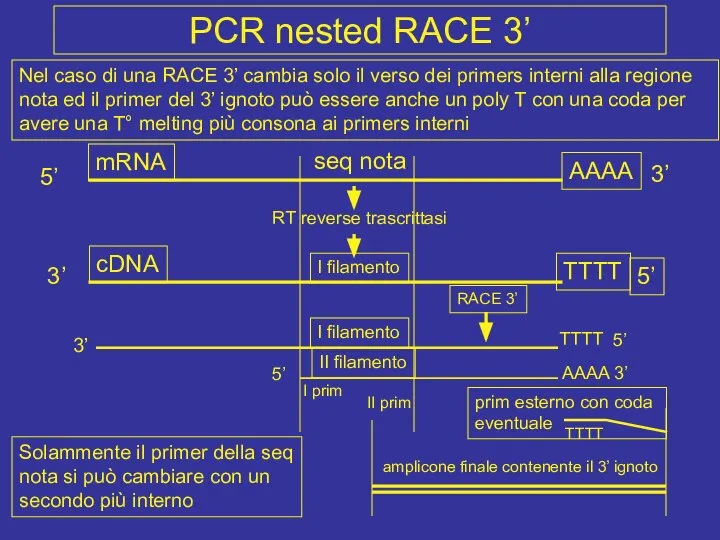
PCR nested RACE 3’
Nel caso di una RACE 3’ cambia solo
PCR nested RACE 3’
Nel caso di una RACE 3’ cambia solo

RACE 5’ Cerchiamo il 5’ ignoto
Dobbiamo comunque ottenere il cDNA ed
RACE 5’ Cerchiamo il 5’ ignoto
Dobbiamo comunque ottenere il cDNA ed

5’
mRNA
poly(A) tail
1 - Annealing tra una regione interna dell’mRNA e
5’
mRNA
poly(A) tail
1 - Annealing tra una regione interna dell’mRNA e

2 - Retrotrascrizione e tailing all’estremità 3’ del cDNA
5’
AAA….AAA
n
5’
3’
Degradazione mRNA
5’
3’
GSP1
Purificazione
2 - Retrotrascrizione e tailing all’estremità 3’ del cDNA
5’
AAA….AAA
n
5’
3’
Degradazione mRNA
5’
3’
GSP1
Purificazione
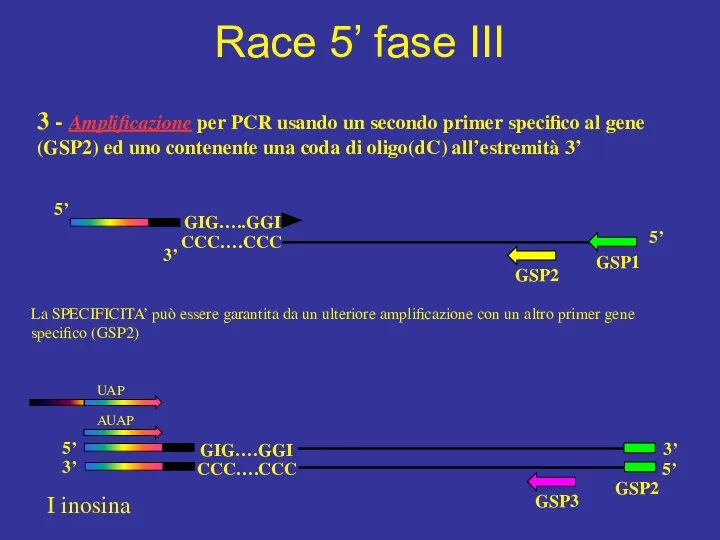
Race 5’ fase III
I inosina
Race 5’ fase III
I inosina
mRNA
poly A
Sintesi del I filamento di cDNA tramite rev.transcript.
primer rev. specifico
5’
3’
5’
3’
5’
3’
cDNA
mRNA
poly A
Sintesi del I filamento di cDNA tramite rev.transcript.
primer rev. specifico
5’
3’
5’
3’
5’
3’
cDNA

dopo la sintesi del I filam. di cDNA e la terminal
dopo la sintesi del I filam. di cDNA e la terminal
 Деление клетки. Митоз. Подготовила учитель химии и биологии Родионова Н.Г. МОБУ «Лобасковская ООШ»
Деление клетки. Митоз. Подготовила учитель химии и биологии Родионова Н.Г. МОБУ «Лобасковская ООШ»  Коммуникация у кошек
Коммуникация у кошек В мире насекомых
В мире насекомых ИММУНИТЕТ от лат. immunitas — освобождение, избавление
ИММУНИТЕТ от лат. immunitas — освобождение, избавление  Одноклеточные ядерные организмы
Одноклеточные ядерные организмы Плесневые грибы и дрожжи
Плесневые грибы и дрожжи Презентация на тему "Рыбы" - скачать бесплатно презентации по Биологии
Презентация на тему "Рыбы" - скачать бесплатно презентации по Биологии Фолдинг белка. Физикохимические свойства белков
Фолдинг белка. Физикохимические свойства белков Деление клетки
Деление клетки Физиология цнс. Промежуточный мозг. Базальные ганглии. Кора больших полушарий головного мозга
Физиология цнс. Промежуточный мозг. Базальные ганглии. Кора больших полушарий головного мозга Презентация на тему "Цветение и опыление растений" - презентации по Биологии
Презентация на тему "Цветение и опыление растений" - презентации по Биологии ДНК в генной инженерии
ДНК в генной инженерии Биотехнологии в пищевой промышленности
Биотехнологии в пищевой промышленности Класс Амфибии (Земноводные)
Класс Амфибии (Земноводные) Роль К.Ф. Фукса в истории города (Комбинированная экскурсия для гостей Казани по местам, связанным с К.Ф.Фуксом на 3 часа) Подгот
Роль К.Ф. Фукса в истории города (Комбинированная экскурсия для гостей Казани по местам, связанным с К.Ф.Фуксом на 3 часа) Подгот Кровообрщение
Кровообрщение Птицы. 7 класс
Птицы. 7 класс Регуляция и патология липидного обмена
Регуляция и патология липидного обмена Многообразие и значение грибов
Многообразие и значение грибов Тема урока: Эволюция кровеносной системы.
Тема урока: Эволюция кровеносной системы. Тип Плоские черви. Вы уже изучили И много нового для себя открыли Симметрия их тела, Скажите без промедления, имеет: Двусторо
Тип Плоские черви. Вы уже изучили И много нового для себя открыли Симметрия их тела, Скажите без промедления, имеет: Двусторо Урок по предмету Байкаловедение. Тема: «Нерпа». 6кл. МОУ СОШ №11 г. Иркутск. Учитель высшей квалификационно
Урок по предмету Байкаловедение. Тема: «Нерпа». 6кл. МОУ СОШ №11 г. Иркутск. Учитель высшей квалификационно Общая характеристика грибов
Общая характеристика грибов Жизненный цикл клетки
Жизненный цикл клетки Презентация на тему "Использование информационно-коммуникационных технологий на уроках биологии" - презентации по Биологии
Презентация на тему "Использование информационно-коммуникационных технологий на уроках биологии" - презентации по Биологии Тип Членистоногие
Тип Членистоногие Популяция. Генетическая единица вида
Популяция. Генетическая единица вида Семейство Астровые или Сложноцветые (Asteraceae или Compositae)
Семейство Астровые или Сложноцветые (Asteraceae или Compositae)