- Болезни клеточных органелл

Содержание
- 2. 1.Митохондриальные болезни
- 3. Митохондрии Митохондриальная ДНК
- 4. Схема строения митохондриальной ДНК
- 5. Углеводы Жиры глюкоза лактат жирные кислоты Ацетил КоА Цикл Кребса Бета-окисление I II III IV V
- 7. Клинические проявления митохондриальных болезней (основные) миопатический синдром - слабость и атрофия мышц, снижение мышечного тонуса, мышечные
- 8. Клинические проявления митохондриальных болезней (дополнительные) снижение слуха сенсоневрального происхождения; нарушение зрения - атрофия зрительных нервов, пигментная
- 9. Схема строения митохондрии
- 10. Феномен RRF (рваных (или шероховатых) красных волокон) в биоптатах мышц при митохондриальных болезнях.
- 12. Синдром Кернса-Сейра
- 13. Синдром Кернса-Сейра Манифестация с 4 - 18 лет Снижение толерантности к физ. нагрузке, миопатич. синдром, птоз,
- 14. Динамика появления клинических симптомов Частичный птоз век - 9 лет Частичная офтальмоплегия - 10,5 лет Снижение
- 15. Причина синдрома Кернса-Сейра - делеция митохондриальной ДНК Размер от 1,3 до 8 тыс. пар нуклеотидов Наиболее
- 16. Делеция митохондриальной ДНК
- 17. Клиническая экспрессия митохондриальной делеции зависит от Локализации делеции в тканях Чувствительности данной ткани к энергетическому дефициту
- 18. Синдром MELAS - (митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды). В основе заболевания – точковая мутация митохондриального гена
- 19. Синдром MERRF (миоклонус-эпилепсия с «рваными» красными волокнами). В основе заболевания – точковая мутация митохондриального гена транспортной
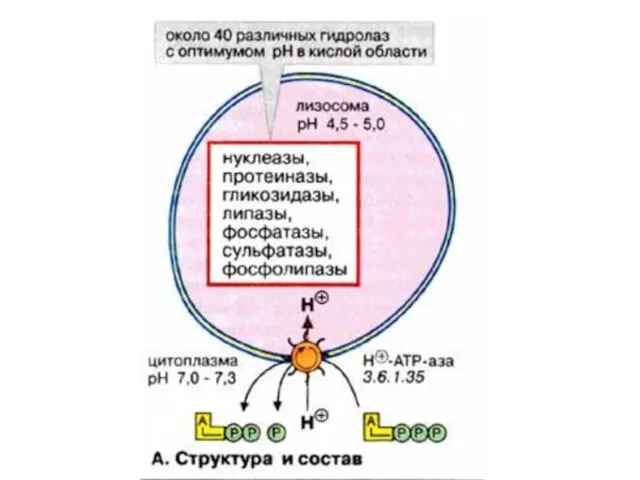
- 20. 2.Лизосомные болезни накопления
- 23. Классификация лизосомных болезней мукополисахаридозы сфинголипидозы муколипидозы
- 24. Мукополисахаридозы Мукополисахариды являются сложными гетерогенными соединениями. Из них хондроитин-серная и гиалуроновые кислоты служат основными строительными элементами
- 25. Лицо больного мукополисахаридозом
- 26. Типы мукополисахаридозов I тип — 1:20000 — 1:25000. Наследование аутосомно-рецессивное. Дефицит α –L – идуронидазы. тип
- 27. Синдром Гурлер (МПСI)
- 28. Синдром Гурлер (МПСI) Ген локализован в 22q11. Клинические признаки: Тяжелая симптоматика и ранняя манифестация. Грубые черты
- 29. Гипертрихоз
- 30. Рентгенологические изменения при синдроме Гурлер: Кубовидной формы позвонки с закругленными контурами, углообразный кифоз пояснично-грудного отдела, гипоплазия
- 31. Синдром Гурлер-Шейе
- 35. Синдром Хантера (МПСII)
- 36. Синдром Хантера (МПСII) Ген локализован в Хq27.1-q28. Клинические признаки: Признаки подобные синдрому Гурлер, но более поздняя
- 39. Ферментозаместительная терапия больных с МПСI и МПСII. Альдуразим разработан американской фирмой «Джинзайм» (GENZYME) для лечения МПСI
- 40. Эффективность лечения ферментозамещающими препаратами «Альдуразим» и «Элапраза» Улучшение общего состояния больных: - нарастание двигательной активности -
- 41. Синдром Моркио (МПСIV)
- 42. Рентгенологические признаки синдрома Моркио: деформация костей таза, вертлужных впадин, гипоплазия головок бедренных костей, деформация костей кистей.
- 46. Морато-Лами синдром (МПСVI)
- 47. 3.Пероксисомные болезни
- 48. Адренолейкодистрофия Дегенеративное заболевание белого вещества головного мозга и надпочечников, обусловленное дефектом обмена жирных кислот. Тип наследования
- 50. Скачать презентацию

1.Митохондриальные болезни
1.Митохондриальные болезни
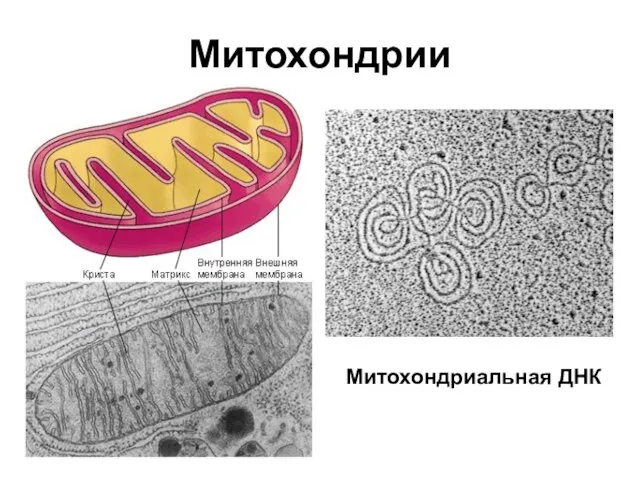
Митохондрии
Митохондриальная ДНК
Митохондрии
Митохондриальная ДНК
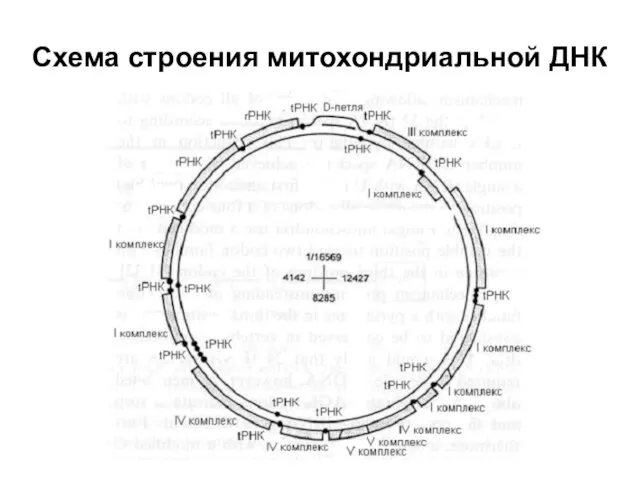
Схема строения митохондриальной ДНК
Схема строения митохондриальной ДНК
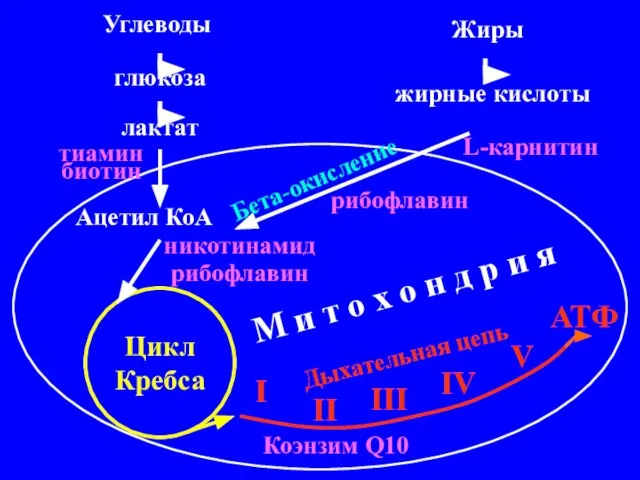
Углеводы
Жиры
глюкоза
лактат
жирные кислоты
Ацетил КоА
Цикл Кребса
Бета-окисление
I
II
III
IV
V
АТФ
Дыхательная цепь
L-карнитин
рибофлавин
никотинамид
рибофлавин
тиамин биотин
Коэнзим Q10
М и т о
Углеводы
Жиры
глюкоза
лактат
жирные кислоты
Ацетил КоА
Цикл Кребса
Бета-окисление
I
II
III
IV
V
АТФ
Дыхательная цепь
L-карнитин
рибофлавин
никотинамид
рибофлавин
тиамин биотин
Коэнзим Q10
М и т о

Клинические проявления митохондриальных болезней (основные)
миопатический синдром - слабость и атрофия мышц,
Клинические проявления митохондриальных болезней (основные)
миопатический синдром - слабость и атрофия мышц,

Клинические проявления митохондриальных болезней (дополнительные)
снижение слуха сенсоневрального происхождения;
нарушение зрения - атрофия
Клинические проявления митохондриальных болезней (дополнительные)
снижение слуха сенсоневрального происхождения;
нарушение зрения - атрофия
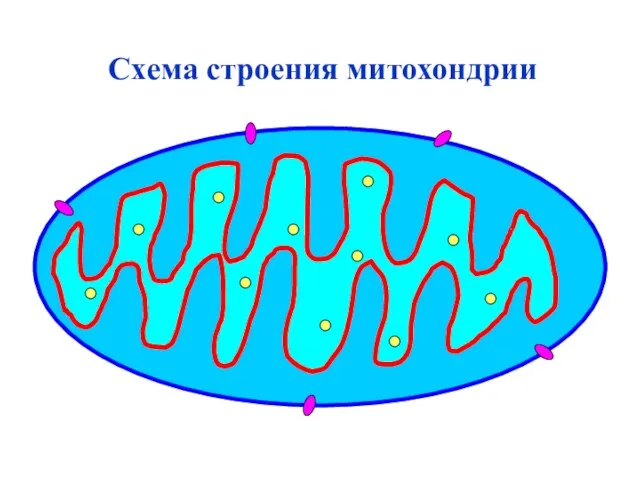
Схема строения митохондрии
Схема строения митохондрии
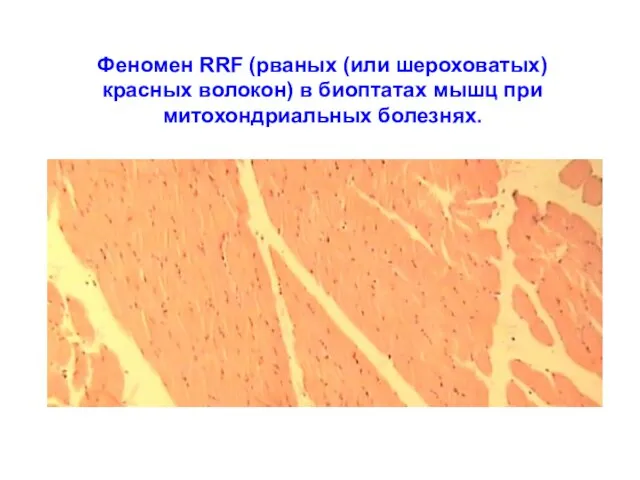
Феномен RRF (рваных (или шероховатых) красных волокон) в биоптатах мышц при
Феномен RRF (рваных (или шероховатых) красных волокон) в биоптатах мышц при
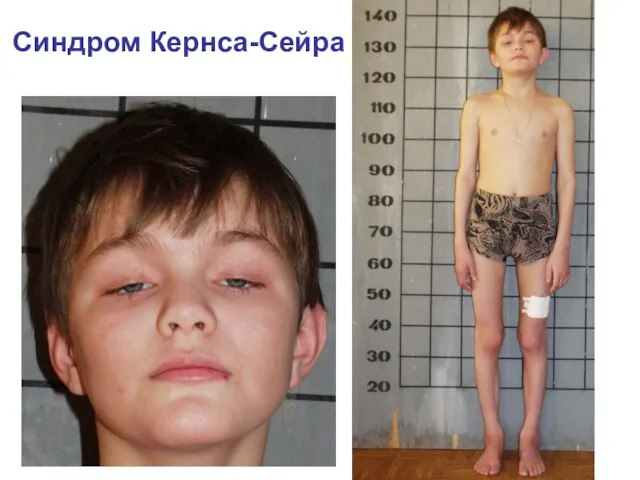
Синдром Кернса-Сейра
Синдром Кернса-Сейра

Синдром Кернса-Сейра
Манифестация с 4 - 18 лет
Снижение толерантности к физ. нагрузке,
Синдром Кернса-Сейра
Манифестация с 4 - 18 лет
Снижение толерантности к физ. нагрузке,

Динамика появления клинических симптомов
Частичный птоз век - 9 лет
Частичная офтальмоплегия -
Динамика появления клинических симптомов
Частичный птоз век - 9 лет
Частичная офтальмоплегия -

Причина синдрома Кернса-Сейра - делеция митохондриальной ДНК
Размер от 1,3 до 8
Причина синдрома Кернса-Сейра - делеция митохондриальной ДНК
Размер от 1,3 до 8
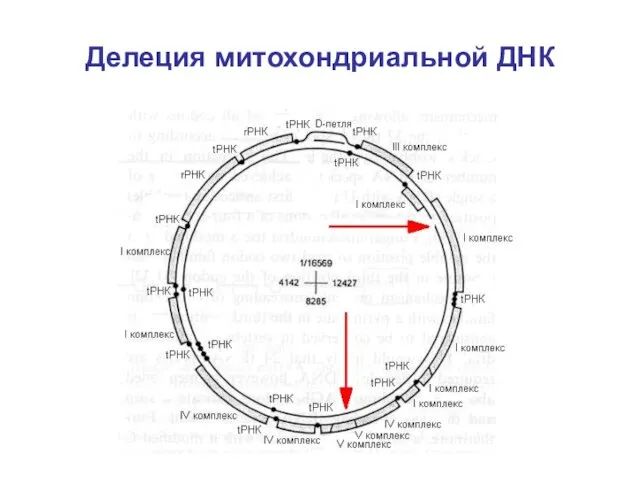
Делеция митохондриальной ДНК
Делеция митохондриальной ДНК

Клиническая экспрессия митохондриальной делеции зависит от
Локализации делеции в тканях
Чувствительности данной ткани
Клиническая экспрессия митохондриальной делеции зависит от
Локализации делеции в тканях
Чувствительности данной ткани
Синдром MELAS -
(митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды).
В основе заболевания – точковая
Синдром MELAS -
(митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды).
В основе заболевания – точковая
Синдром MERRF
(миоклонус-эпилепсия с «рваными» красными волокнами).
В основе заболевания – точковая мутация
Синдром MERRF
(миоклонус-эпилепсия с «рваными» красными волокнами).
В основе заболевания – точковая мутация

2.Лизосомные болезни накопления
2.Лизосомные болезни накопления
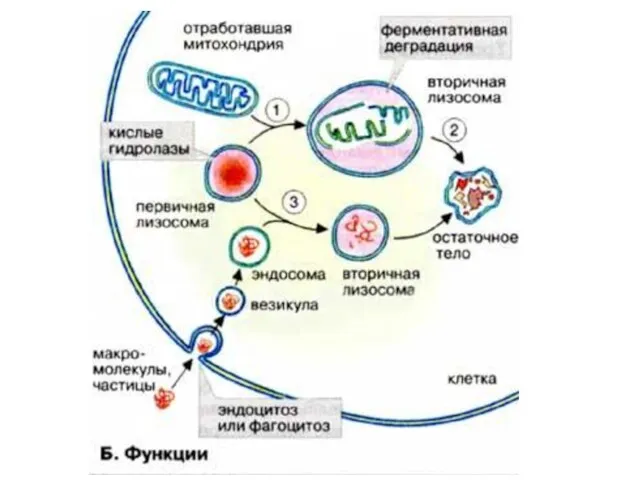

Классификация лизосомных болезней
мукополисахаридозы
сфинголипидозы
муколипидозы
Классификация лизосомных болезней
мукополисахаридозы
сфинголипидозы
муколипидозы

Мукополисахаридозы
Мукополисахариды являются сложными гетерогенными соединениями. Из них хондроитин-серная и гиалуроновые кислоты
Мукополисахаридозы
Мукополисахариды являются сложными гетерогенными соединениями. Из них хондроитин-серная и гиалуроновые кислоты

Лицо больного мукополисахаридозом
Лицо больного мукополисахаридозом

Типы мукополисахаридозов
I тип — 1:20000 — 1:25000. Наследование аутосомно-рецессивное. Дефицит α
Типы мукополисахаридозов
I тип — 1:20000 — 1:25000. Наследование аутосомно-рецессивное. Дефицит α
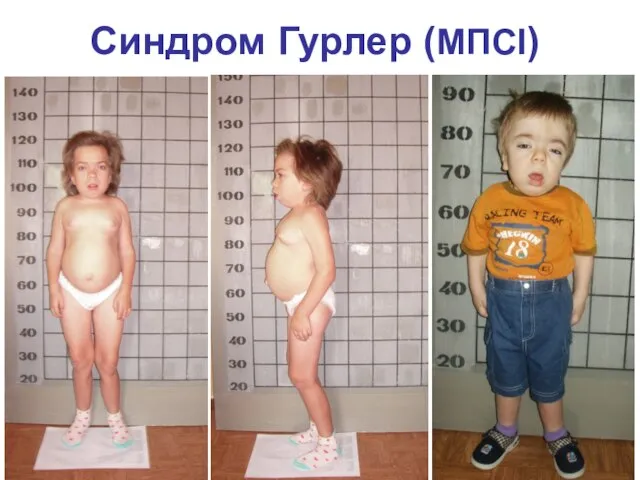
Синдром Гурлер (МПСI)
Синдром Гурлер (МПСI)

Синдром Гурлер (МПСI)
Ген локализован в 22q11.
Клинические признаки:
Тяжелая симптоматика и ранняя
Синдром Гурлер (МПСI)
Ген локализован в 22q11.
Клинические признаки:
Тяжелая симптоматика и ранняя
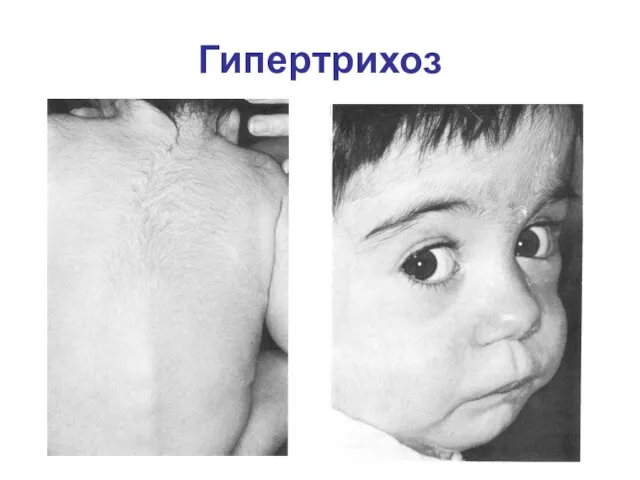
Гипертрихоз
Гипертрихоз
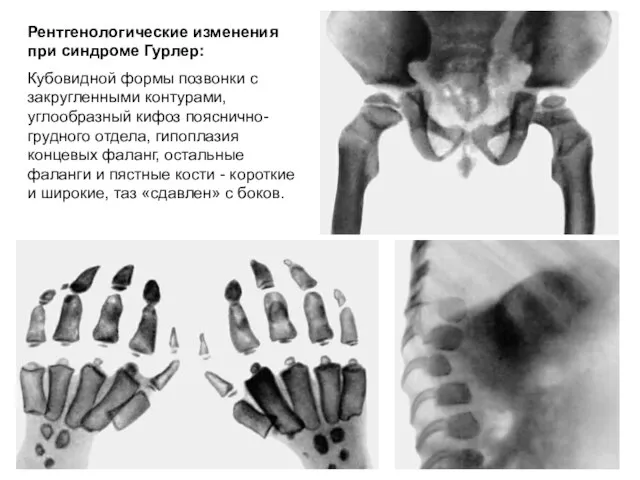
Рентгенологические изменения при синдроме Гурлер:
Кубовидной формы позвонки с закругленными контурами,
Рентгенологические изменения при синдроме Гурлер:
Кубовидной формы позвонки с закругленными контурами,
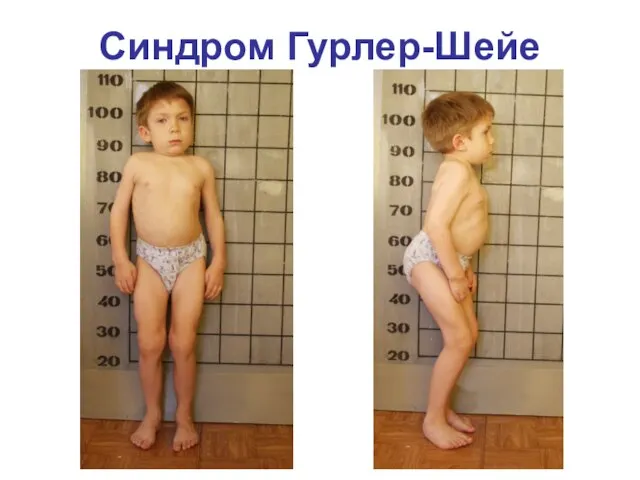
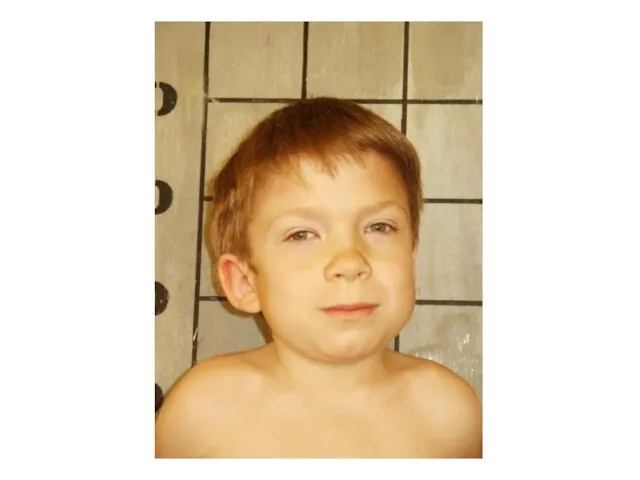
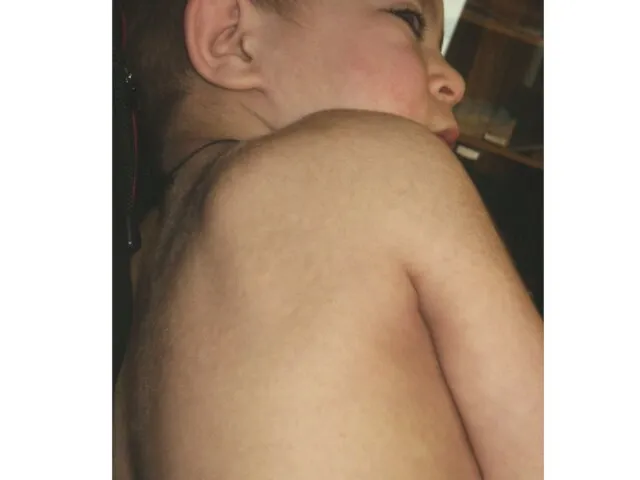
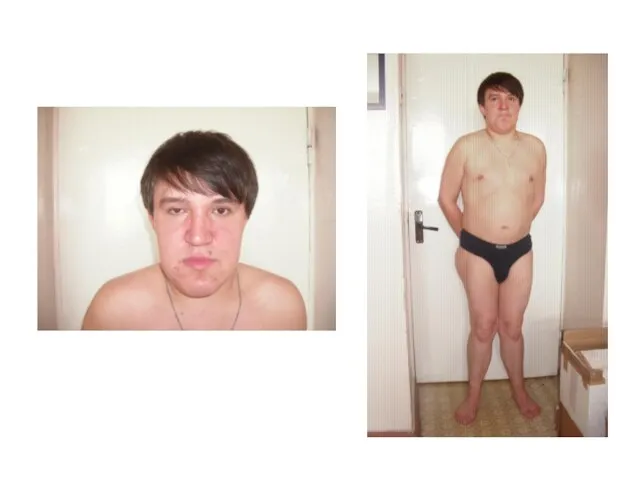
Синдром Гурлер-Шейе
Синдром Гурлер-Шейе



Синдром Хантера (МПСII)
Синдром Хантера (МПСII)

Синдром Хантера (МПСII)
Ген локализован в Хq27.1-q28.
Клинические признаки:
Признаки подобные синдрому
Синдром Хантера (МПСII)
Ген локализован в Хq27.1-q28.
Клинические признаки:
Признаки подобные синдрому

Ферментозаместительная терапия больных с МПСI и МПСII.
Альдуразим разработан американской фирмой
Ферментозаместительная терапия больных с МПСI и МПСII.
Альдуразим разработан американской фирмой

Эффективность лечения ферментозамещающими препаратами «Альдуразим» и «Элапраза»
Улучшение общего состояния больных:
-
Эффективность лечения ферментозамещающими препаратами «Альдуразим» и «Элапраза»
Улучшение общего состояния больных:
-

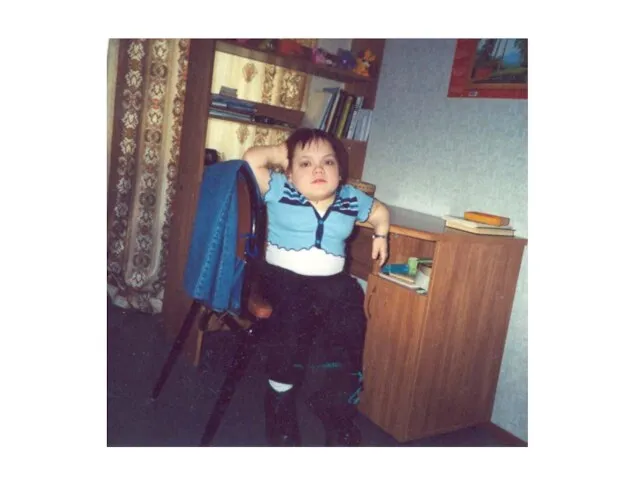
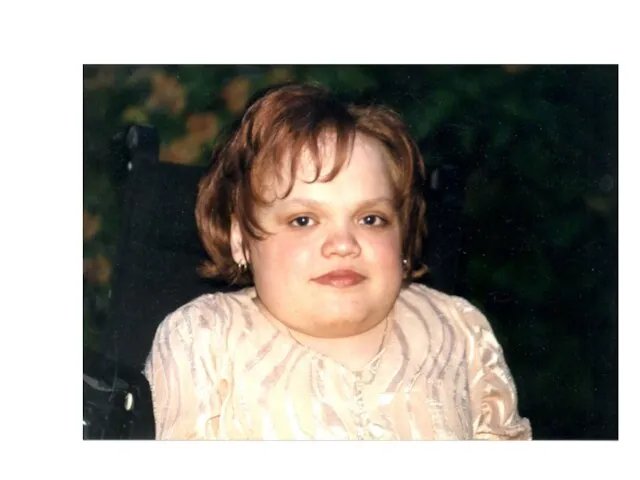
Синдром Моркио (МПСIV)
Синдром Моркио (МПСIV)
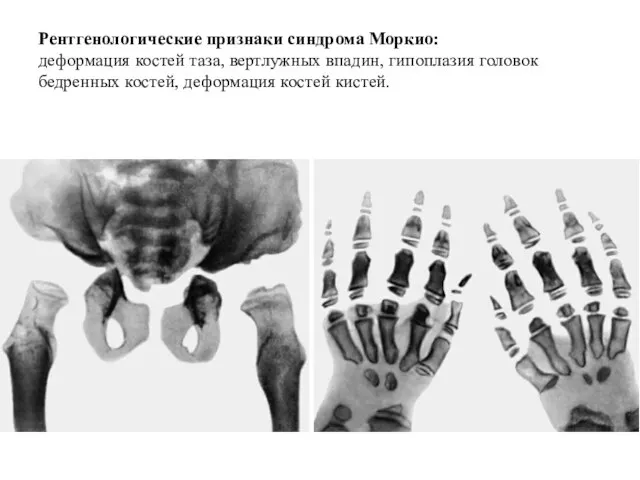
Рентгенологические признаки синдрома Моркио:
деформация костей таза, вертлужных впадин, гипоплазия головок бедренных
Рентгенологические признаки синдрома Моркио: деформация костей таза, вертлужных впадин, гипоплазия головок бедренных

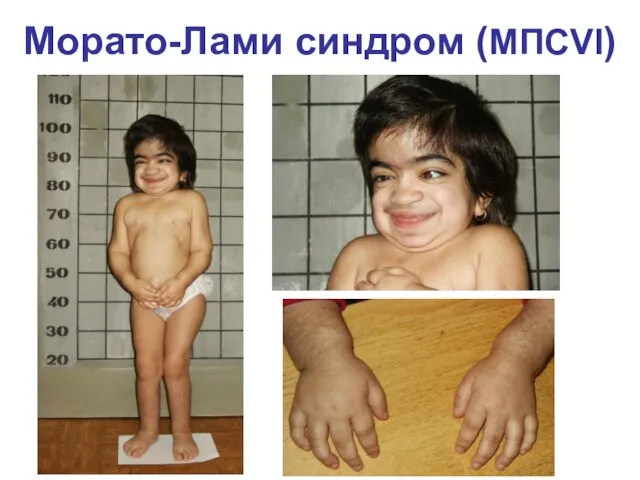
Морато-Лами синдром (МПСVI)
Морато-Лами синдром (МПСVI)

3.Пероксисомные болезни
3.Пероксисомные болезни

Адренолейкодистрофия
Дегенеративное заболевание белого вещества головного мозга и надпочечников, обусловленное дефектом обмена
Адренолейкодистрофия
Дегенеративное заболевание белого вещества головного мозга и надпочечников, обусловленное дефектом обмена
 Балалардың керек-жарақтарына, жиһаздарына қойылатын гигиеналық талаптар
Балалардың керек-жарақтарына, жиһаздарына қойылатын гигиеналық талаптар Фармакология психотроптных средств возбуждающего типа действия
Фармакология психотроптных средств возбуждающего типа действия Итоги работы онкологического отделения № 2 за 2021 год
Итоги работы онкологического отделения № 2 за 2021 год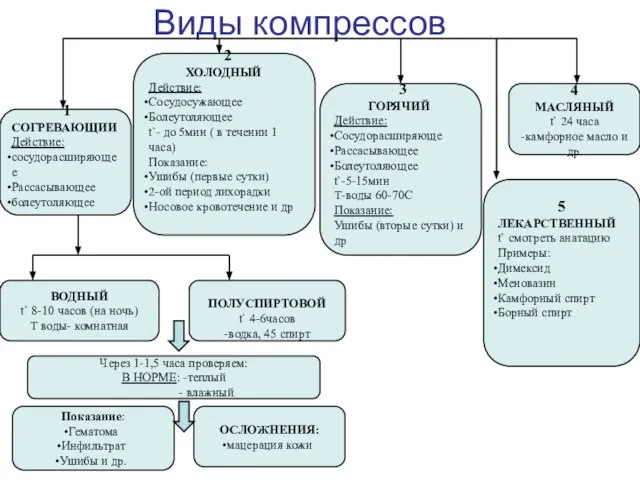 Виды компрессов
Виды компрессов Заболевания детей раннего возраста. Хронические расстройства питания
Заболевания детей раннего возраста. Хронические расстройства питания Оказание первой доврачебной помощи пострадавшим
Оказание первой доврачебной помощи пострадавшим Адаптація новонароджених дітей. Особливості надання допомоги недоношеним дітям
Адаптація новонароджених дітей. Особливості надання допомоги недоношеним дітям Фармацевтическая несовместимость ингредиентов в лекарственных препаратах индивидуального изготовления
Фармацевтическая несовместимость ингредиентов в лекарственных препаратах индивидуального изготовления Портальная гипертензия
Портальная гипертензия Балалар жасындағы терапиялық стоматологияның даму сатылары
Балалар жасындағы терапиялық стоматологияның даму сатылары Перинатальное поражение нервной системы. Детский церебральный паралич. Минимальная мозговая дисфункция
Перинатальное поражение нервной системы. Детский церебральный паралич. Минимальная мозговая дисфункция Основы иммуногематологии
Основы иммуногематологии Понятие о предболезни, выявление предболезни надгрузочнымыми пробами
Понятие о предболезни, выявление предболезни надгрузочнымыми пробами Ультразвуковое исследование. Ультразвуковая диагностика в педиатрии
Ультразвуковое исследование. Ультразвуковая диагностика в педиатрии Преэкламсиясы бар жүкті әйелдерде
Преэкламсиясы бар жүкті әйелдерде Сердечно-сосудистая система. Искусственные клапаны сердца
Сердечно-сосудистая система. Искусственные клапаны сердца Плешкова 3 тема
Плешкова 3 тема Основные причины приобретенных клапанных пороков сердца у детей
Основные причины приобретенных клапанных пороков сердца у детей Гигиена водоснабжения
Гигиена водоснабжения Естетичні операції на грудях, животі, кінцівках. Інші види косметичних втручань
Естетичні операції на грудях, животі, кінцівках. Інші види косметичних втручань Профилактика стоматологических заболеваний
Профилактика стоматологических заболеваний Хронический пульпит
Хронический пульпит Наиболее частые осложнения от наркоза
Наиболее частые осложнения от наркоза Портальная гипертензия
Портальная гипертензия Абьюзивные отношения
Абьюзивные отношения Первичная и вторичная профилактика депрессий у лиц пожилого возраста на базе геронтологического центра
Первичная и вторичная профилактика депрессий у лиц пожилого возраста на базе геронтологического центра Химиотерапия рака легких
Химиотерапия рака легких Das Gefäßzentrum Niederlausitz Igor Grobel 20. Symposium der Vereinigung der Gefäßchirurgen Brandenburgs Bad Saarow
Das Gefäßzentrum Niederlausitz Igor Grobel 20. Symposium der Vereinigung der Gefäßchirurgen Brandenburgs Bad Saarow