- Характеристика некоторых форм первичных иммунодефицитов

Содержание
- 2. «Характеристика некоторых форм первичных иммунодефицитов». Цикл 2 - клиническая иммунология Занятие № 3
- 3. Фронтальный опрос -вопросы Дайте определение понятию первичная иммунологическая недостаточность. Назовите основные клетки врожденного иммунитета и их
- 4. Характеристика некоторых форм ПИД Тяжелый комбинированный иммунодефицит (ТКИД). Болезнь Брутона. Общая вариабельная иммунная недостаточность (ОВИН). Селективный
- 5. Тяжелый комбинированный иммунодефицит (ТКИД) Возраст: развивается у детей младше 2-х лет. Генетичекий дефект: выявлено 93 патогенных
- 6. ТКИН, Х-сцепленный Мутация р.Cys182Tyr в экзоне 4 гена IL2RG.
- 7. КЛАССИФИКАЦИЯ СИНДРОМОВ ТКИН Тяжелый комбинированные иммунодефициты
- 8. ИСТОРИЯ “BUBBLE-BOY”
- 9. ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ИММУНОДЕФИЦИТЫ Клинические проявления развиваются с первых недель жизни: Персистирующая диарея Инфекции респираторного тракта Оппортунистические
- 10. ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ММУНОДЕФИЦИТЫ Терапия: Единственным методом терапии ТКИН является трансплантация костного мозга. При наличии HLA-идентичного родственного
- 11. Болезнь Брутона: описана в 1952 году Возраст: проявления у детей с первого полугодия жизни (когда заканчивается
- 12. Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов. Агаммаглобулинемия. Костный мозг Периферия Про В Переклю чение изотипов
- 13. Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов. Агаммаглобулинемия. мутация в CD-19
- 14. АГАММАГЛОБУЛИНЕМИЯ С ДЕФИЦИТОМ В-КЛЕТОК Клинические проявления ИНФЕКЦИОННЫЕ Рецидивирующие бактериальные инфекции: ЛОР-органов, легких, ЖКТ, кожи Менингиты, артриты,
- 15. Клинические примеры: агаммаглобулинемия с дефицитом В-клеток (Гусева М. Н.,Семенов А. В.,2014) Пациенты: однояйцевые сибсы Ш.А. и
- 16. Данные лабораторного обследования (Гусева М. Н.,Семенов А. В.,2014) Ш.В., 2008 г.р. Ш.А., 2008 г.р.
- 18. Поствакцинальные антитела (Гусева М. Н.,Семенов А. В.,2014)
- 19. Генетический анализ Проведен генетический анализ образца крови методами ПЦР и SSCP с последующим секвенированием фрагментов (19
- 20. Схема введения в/в иммуноглобулинов (октагам, 5%) Недели наблюдения Концентрация IgG, г/л Насыщение – 4 недели, далее
- 21. Общая вариабельная иммунная недостаточность (ОВИН) Возраст: начало болезни в возрасте старше 2-х лет. Плохой ответ на
- 22. ОВИН: мутации в генах , кодирующих белки, вовлеченные в В-лимфопоэз и гомеостаз В-клеток (включая CD 19
- 23. Селективный иммунодефицит IgA Возраст: встречается у пациентов старше 4 лет. Генетичекий дефект: результат генетического дефекта: снижение
- 24. Гипер-IgE-синдром Характеристики: в крови существенно повышены уровни иммуноглобулина класса Е. Возраст: врожденные аномалии строения лицевого скелета.
- 25. Гипер-IgE-синдром Описаны также мутации в гене, кодирующем STAT 3 (аутосомно-доминантное наследование). Результат –полиорганная патология. Клиника: повторные
- 26. Транзиторная агаммаглобулинемия Возраст: диагностируется у пациентов в возрасте от 6 месяцев до 2-3 лет. Генетичекий дефект:
- 27. Синдром Ди-Джорджи Возраст: с рождения - гипоплазия или аплазия тимуса, множественные дефекты артериального ствола, Генетичекий дефект:
- 28. Алгоритм ультразвукового исследования тимуса осмотр перешейка тимуса левой доли правой доли
- 29. Синдром Ди-Джорджи Результат генетического дефекта: стволовые клетки не дифференцируются в Т-лимфоциты. Т-иммунодефицит: число и активность Т
- 30. Атаксия –телеангиоэктазия (синдром Луи-Барр) Характеристики: прогрессирующая мозжечковая атаксия, сочетающаяся с задержкой роста и отсутствием иммуноглобулинов классов
- 31. Атаксия –телеангиоэктазия (синдром Луи-Барр) Характеристики: прогрессирующая мозжечковая атаксия, сочетающаяся с задержкой роста и отсутствием иммуноглобулинов классов
- 32. Атаксия –телеангиоэктазия (синдром Луи-Барр) Атаксическая походка (мозжечковая недостаточность прогрессирует)
- 33. Синдром Вискотт-Олдриджа Характеристики: тромбоцитопения. Врожденные аномалии тромбоцитарного звена: резко выраженное изменение морфологии тромбоцитов и существенно сниженное
- 34. Хронический кожно-слизистый кандидоз Генетичекий дефект: не установлен. Характерны: частые поражения Candida Albicans кожи и слизистых оболочек.
- 35. Врожденные дефекты фагоцитоза В настоящее время выделяют более 20 нозологических форм. К ним относятся дефекты числа
- 36. Хроническая гранулематозная болезнь (ХГБ) Характеристики: фагоциты не способны генерировать радикалы кислорода, нарушена способность к «кислородному взрыву».
- 37. Хроническая гранулематозная болезнь (ХГБ) Характерны: рецидивирующие инфекционные заболевания бронхолегочной системы, поражающие также кожу, лимфатические узлы, печень,
- 38. Хроническая гранулематозная болезнь (Клинический пример) (Гусева М. Н., Семенов А. В.,2014) Б.М., 2010 г.р. Ребенок от
- 39. Хроническая гранулематозная болезнь (Клинический пример) (Гусева М. Н., Семенов А. В., 2014) В 1 год –
- 40. Клинический анализ крови с 23.10.12 по 20.11.12
- 41. Содержание иммуноглобулинов в сыворотке крови в 2 г. 3 мес.
- 42. Результаты Burst-теста
- 43. Дефекты рецепторов и сигнальных компонентов врожденного иммунитета Известны ПИД с нарушениями в TLR-сигнальной системе, вызванные дефектами:
- 44. Типы иммунного ответа
- 45. Типы иммунного ответа
- 46. Аутовоспалительные синдромы, связанные с дисфункцией врожденного иммунитета за счет изменений в системе NOD-подобных рецепторов Группа заболеваний,
- 47. Дефициты системы комплемента Редкие формы ПИД (в подавляющем большинстве случаев дефекты системы комплемента являются вторичными). В
- 48. Особенности проявления симптомов ПИД
- 49. Сруктура летальности ПИД
- 50. Вопросы
- 51. Тестовые вопросы Изолированное снижение уровня IgA в сыворотке крови определяют при: ТКИД. Транзиторной гипогаммаглобулинемии. Общей вариабельной
- 52. Тестовые вопросы Транзиторная гипогаммаглобулинемия диагностируется в возрасте: С 6 месяцев до 3 лет. С рождения до
- 53. Тестовые вопросы При аутовоспалительных нарушениях имеет место дисфункция врожденного иммунитета за счет изменения в системе NOD-подобных
- 54. Тестовые вопросы С наибольшей частотой в популяции встречается: Кожно-слизистый кандидоз Транзиторная гипогаммаглобулинемия. Общая вариабельная иммунная недостаточность
- 55. Тестовые вопросы К врожденным дефектам фагоцитарного звена относятся: Нарушение хемотаксиса. Нарушения адгезии. Нарушение эндоцитоза. Нарушение киллинга.
- 59. Скачать презентацию

«Характеристика некоторых форм первичных иммунодефицитов».
Цикл 2 - клиническая иммунология
Занятие № 3
«Характеристика некоторых форм первичных иммунодефицитов».
Цикл 2 - клиническая иммунология
Занятие № 3

Фронтальный опрос -вопросы
Дайте определение понятию первичная иммунологическая недостаточность.
Назовите основные клетки врожденного
Фронтальный опрос -вопросы
Дайте определение понятию первичная иммунологическая недостаточность.
Назовите основные клетки врожденного

Характеристика некоторых форм ПИД
Тяжелый комбинированный иммунодефицит (ТКИД).
Болезнь Брутона.
Общая вариабельная иммунная недостаточность
Характеристика некоторых форм ПИД
Тяжелый комбинированный иммунодефицит (ТКИД).
Болезнь Брутона.
Общая вариабельная иммунная недостаточность

Тяжелый комбинированный иммунодефицит (ТКИД)
Возраст: развивается у детей младше 2-х лет.
Генетичекий дефект:
Тяжелый комбинированный иммунодефицит (ТКИД)
Возраст: развивается у детей младше 2-х лет.
Генетичекий дефект:
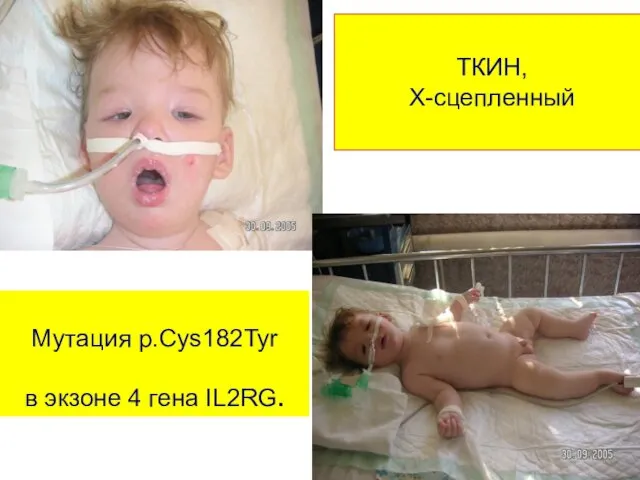
ТКИН,
Х-сцепленный
Мутация р.Cys182Tyr
в экзоне 4 гена IL2RG.
ТКИН,
Х-сцепленный
Мутация р.Cys182Tyr
в экзоне 4 гена IL2RG.
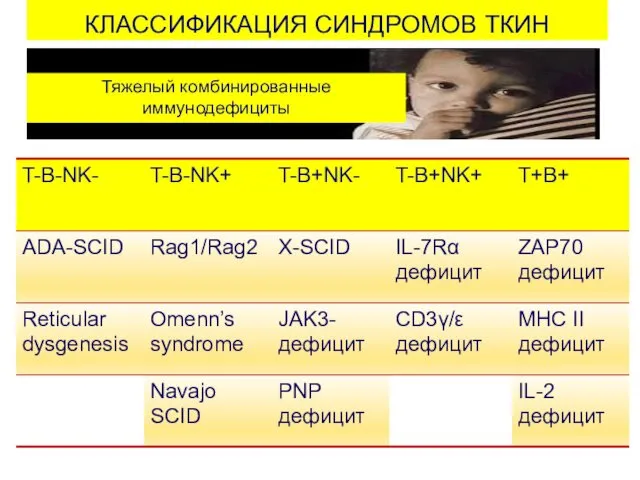
КЛАССИФИКАЦИЯ СИНДРОМОВ ТКИН
Тяжелый комбинированные
иммунодефициты
Тяжелый комбинированные
иммунодефициты

ИСТОРИЯ “BUBBLE-BOY”
ИСТОРИЯ “BUBBLE-BOY”
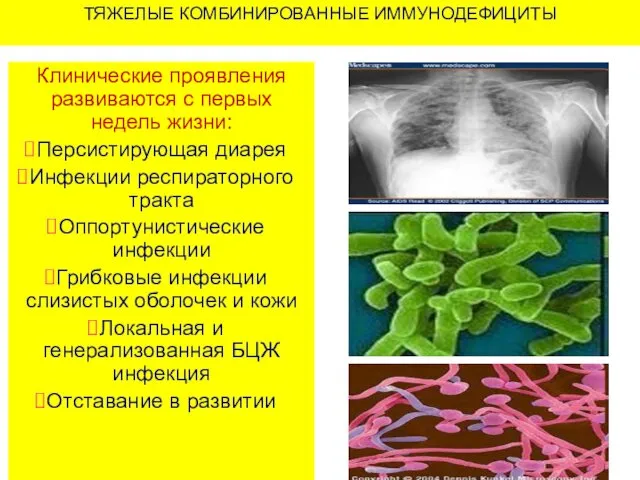
ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ИММУНОДЕФИЦИТЫ
Клинические проявления развиваются с первых недель жизни:
Персистирующая диарея
Инфекции респираторного
ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ИММУНОДЕФИЦИТЫ
Клинические проявления развиваются с первых недель жизни:
Персистирующая диарея
Инфекции респираторного

ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ММУНОДЕФИЦИТЫ
Терапия:
Единственным методом терапии ТКИН является трансплантация костного мозга.
При наличии
ТЯЖЕЛЫЕ КОМБИНИРОВАННЫЕ ММУНОДЕФИЦИТЫ
Терапия:
Единственным методом терапии ТКИН является трансплантация костного мозга.
При наличии

Болезнь Брутона: описана в 1952 году
Возраст: проявления у детей с первого
Болезнь Брутона: описана в 1952 году
Возраст: проявления у детей с первого
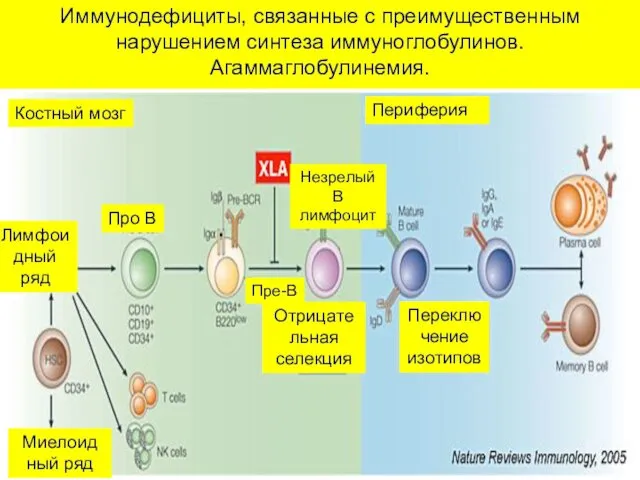
Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов.
Агаммаглобулинемия.
Костный мозг
Периферия
Про В
Переклю
чение изотипов
Пре-В
Миелоид
ный ряд
Лимфоидный
Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов.
Агаммаглобулинемия.
Костный мозг
Периферия
Про В
Переклю
чение изотипов
Пре-В
Миелоид
ный ряд
Лимфоидный

Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов.
Агаммаглобулинемия.
мутация в
CD-19
Иммунодефициты, связанные с преимущественным нарушением синтеза иммуноглобулинов.
Агаммаглобулинемия.
мутация в
CD-19

АГАММАГЛОБУЛИНЕМИЯ С ДЕФИЦИТОМ В-КЛЕТОК
Клинические проявления
ИНФЕКЦИОННЫЕ
Рецидивирующие бактериальные инфекции: ЛОР-органов, легких, ЖКТ, кожи
АГАММАГЛОБУЛИНЕМИЯ С ДЕФИЦИТОМ В-КЛЕТОК
Клинические проявления
ИНФЕКЦИОННЫЕ
Рецидивирующие бактериальные инфекции: ЛОР-органов, легких, ЖКТ, кожи
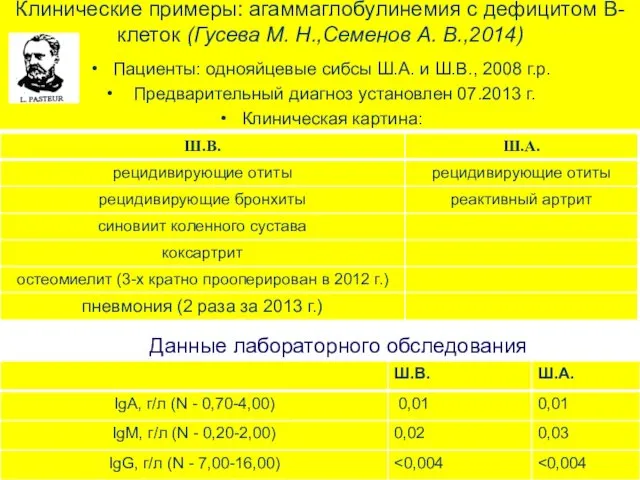
Клинические примеры: агаммаглобулинемия с дефицитом В-клеток (Гусева М. Н.,Семенов А. В.,2014)
Пациенты:
Клинические примеры: агаммаглобулинемия с дефицитом В-клеток (Гусева М. Н.,Семенов А. В.,2014)
Пациенты:

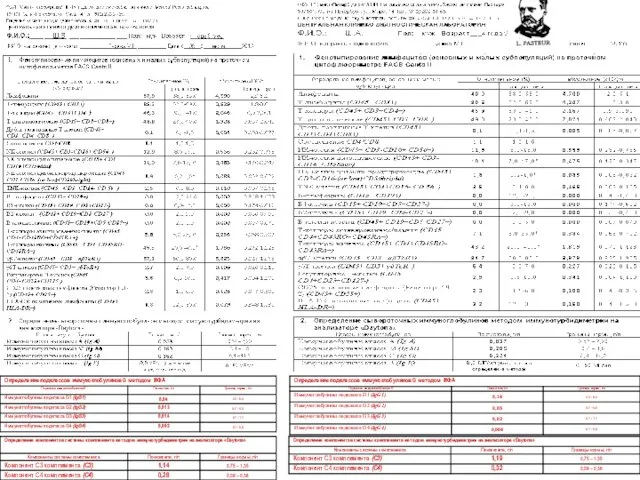
Данные лабораторного обследования
(Гусева М. Н.,Семенов А. В.,2014)
Ш.В., 2008 г.р.
Ш.А., 2008 г.р.
Данные лабораторного обследования
(Гусева М. Н.,Семенов А. В.,2014)
Ш.В., 2008 г.р.
Ш.А., 2008 г.р.
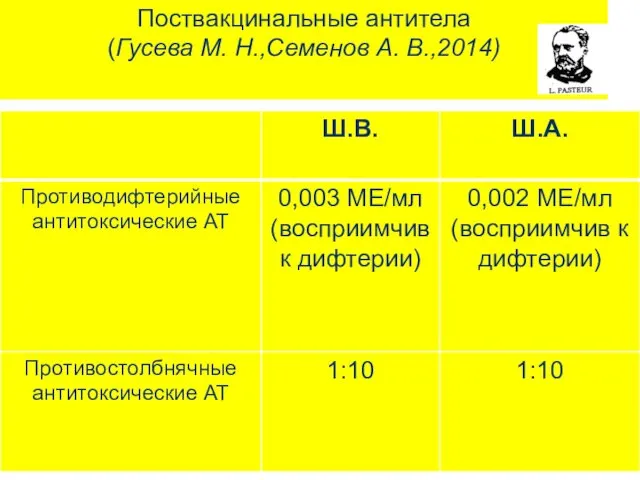
Поствакцинальные антитела
(Гусева М. Н.,Семенов А. В.,2014)
Поствакцинальные антитела
(Гусева М. Н.,Семенов А. В.,2014)

Генетический анализ
Проведен генетический анализ образца крови методами ПЦР и SSCP с
Генетический анализ
Проведен генетический анализ образца крови методами ПЦР и SSCP с

Схема введения в/в иммуноглобулинов (октагам, 5%)
Недели наблюдения
Концентрация IgG, г/л
Насыщение – 4
Схема введения в/в иммуноглобулинов (октагам, 5%)
Недели наблюдения
Концентрация IgG, г/л
Насыщение – 4

Общая вариабельная иммунная недостаточность (ОВИН)
Возраст: начало болезни в возрасте старше 2-х
Общая вариабельная иммунная недостаточность (ОВИН)
Возраст: начало болезни в возрасте старше 2-х

ОВИН: мутации в генах , кодирующих белки, вовлеченные в В-лимфопоэз и
ОВИН: мутации в генах , кодирующих белки, вовлеченные в В-лимфопоэз и

Селективный иммунодефицит IgA
Возраст: встречается у пациентов старше 4 лет.
Генетичекий дефект: результат
Селективный иммунодефицит IgA
Возраст: встречается у пациентов старше 4 лет.
Генетичекий дефект: результат
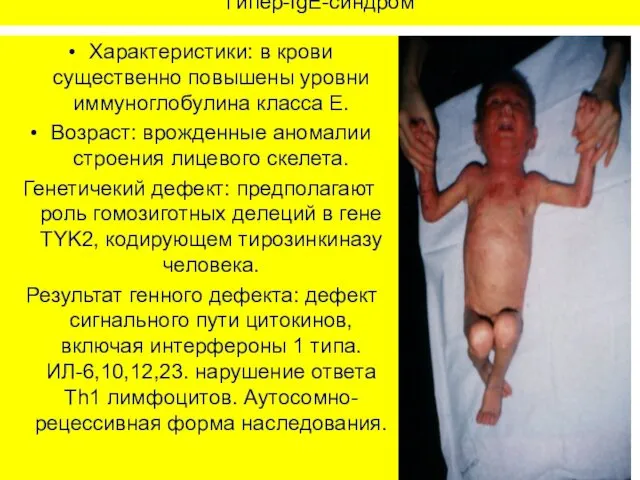
Гипер-IgE-синдром
Характеристики: в крови существенно повышены уровни иммуноглобулина класса Е.
Возраст: врожденные аномалии
Гипер-IgE-синдром
Характеристики: в крови существенно повышены уровни иммуноглобулина класса Е.
Возраст: врожденные аномалии

Гипер-IgE-синдром
Описаны также мутации в гене, кодирующем STAT 3 (аутосомно-доминантное наследование). Результат
Гипер-IgE-синдром
Описаны также мутации в гене, кодирующем STAT 3 (аутосомно-доминантное наследование). Результат

Транзиторная агаммаглобулинемия
Возраст: диагностируется у пациентов в возрасте от 6 месяцев до
Транзиторная агаммаглобулинемия
Возраст: диагностируется у пациентов в возрасте от 6 месяцев до

Синдром Ди-Джорджи
Возраст: с рождения - гипоплазия или аплазия тимуса, множественные дефекты
Синдром Ди-Джорджи
Возраст: с рождения - гипоплазия или аплазия тимуса, множественные дефекты
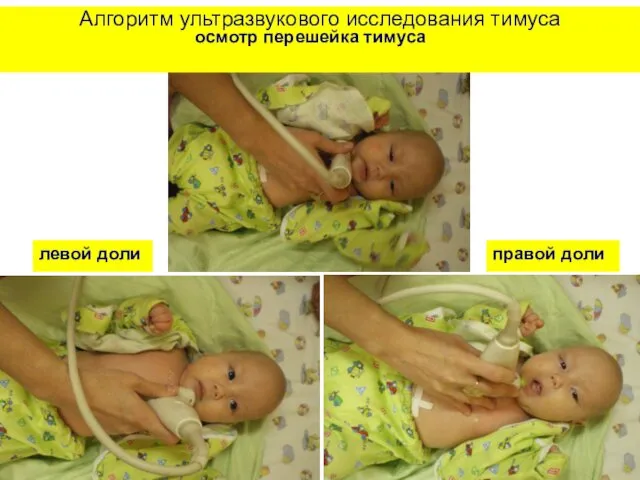
Алгоритм ультразвукового исследования тимуса
осмотр перешейка тимуса
левой доли
правой доли
Алгоритм ультразвукового исследования тимуса
осмотр перешейка тимуса
левой доли
правой доли

Синдром Ди-Джорджи
Результат генетического дефекта: стволовые клетки не дифференцируются в Т-лимфоциты.
Синдром Ди-Джорджи
Результат генетического дефекта: стволовые клетки не дифференцируются в Т-лимфоциты.

Атаксия –телеангиоэктазия (синдром Луи-Барр)
Характеристики: прогрессирующая мозжечковая атаксия, сочетающаяся с задержкой роста
Атаксия –телеангиоэктазия (синдром Луи-Барр)
Характеристики: прогрессирующая мозжечковая атаксия, сочетающаяся с задержкой роста

Атаксия –телеангиоэктазия (синдром Луи-Барр)
Характеристики: прогрессирующая мозжечковая атаксия, сочетающаяся с задержкой роста
Атаксия –телеангиоэктазия (синдром Луи-Барр)
Характеристики: прогрессирующая мозжечковая атаксия, сочетающаяся с задержкой роста

Атаксия –телеангиоэктазия (синдром Луи-Барр)
Атаксическая походка (мозжечковая недостаточность прогрессирует)
Атаксия –телеангиоэктазия (синдром Луи-Барр)
Атаксическая походка (мозжечковая недостаточность прогрессирует)

Синдром Вискотт-Олдриджа
Характеристики: тромбоцитопения.
Врожденные аномалии тромбоцитарного звена: резко выраженное изменение морфологии
Синдром Вискотт-Олдриджа
Характеристики: тромбоцитопения.
Врожденные аномалии тромбоцитарного звена: резко выраженное изменение морфологии
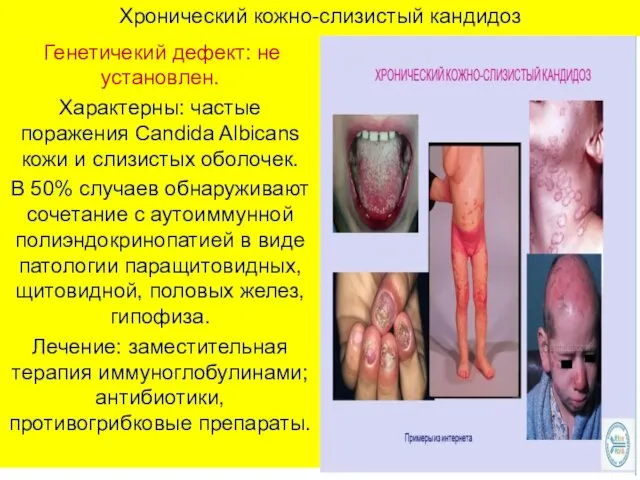
Хронический кожно-слизистый кандидоз
Генетичекий дефект: не установлен.
Характерны: частые поражения Candida Albicans
Хронический кожно-слизистый кандидоз
Генетичекий дефект: не установлен.
Характерны: частые поражения Candida Albicans

Врожденные дефекты фагоцитоза
В настоящее время выделяют более 20 нозологических форм.
К ним
Врожденные дефекты фагоцитоза
В настоящее время выделяют более 20 нозологических форм.
К ним

Хроническая гранулематозная болезнь (ХГБ)
Характеристики: фагоциты не способны генерировать радикалы кислорода, нарушена
Хроническая гранулематозная болезнь (ХГБ)
Характеристики: фагоциты не способны генерировать радикалы кислорода, нарушена

Хроническая гранулематозная болезнь (ХГБ)
Характерны: рецидивирующие инфекционные заболевания бронхолегочной системы, поражающие также
Хроническая гранулематозная болезнь (ХГБ)
Характерны: рецидивирующие инфекционные заболевания бронхолегочной системы, поражающие также

Хроническая гранулематозная болезнь
(Клинический пример) (Гусева М. Н.,
Семенов А. В.,2014)
Б.М., 2010 г.р.
Хроническая гранулематозная болезнь
(Клинический пример) (Гусева М. Н.,
Семенов А. В.,2014)
Б.М., 2010 г.р.

Хроническая гранулематозная болезнь
(Клинический пример) (Гусева М. Н., Семенов А. В.,
2014)
В 1
Хроническая гранулематозная болезнь
(Клинический пример) (Гусева М. Н., Семенов А. В.,
2014)
В 1

Клинический анализ крови
с 23.10.12 по 20.11.12
Клинический анализ крови
с 23.10.12 по 20.11.12
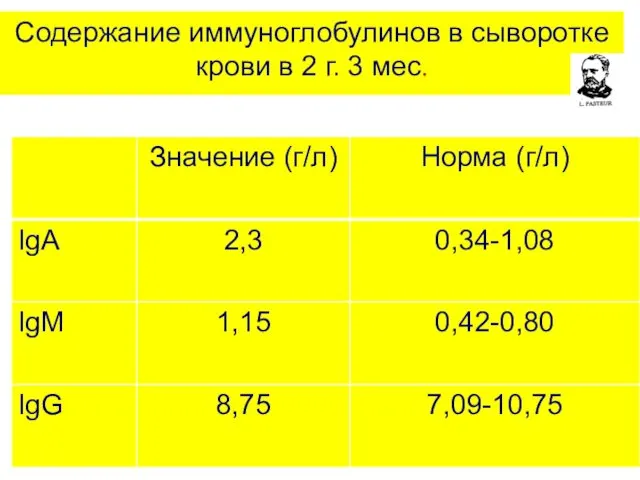
Содержание иммуноглобулинов в сыворотке крови в 2 г. 3 мес.
Содержание иммуноглобулинов в сыворотке крови в 2 г. 3 мес.
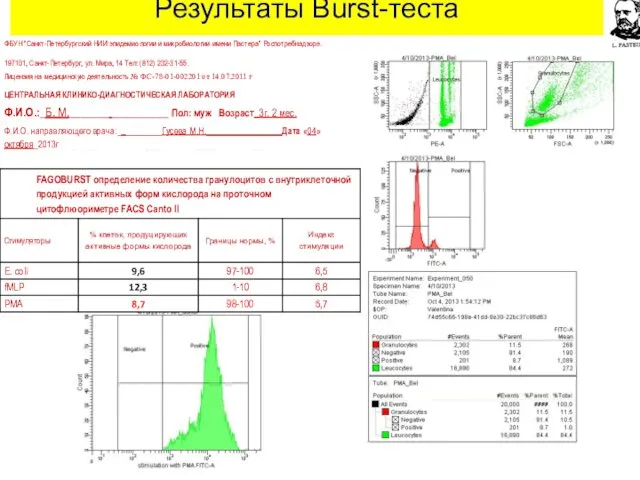
Результаты Burst-теста
Результаты Burst-теста

Дефекты рецепторов и сигнальных компонентов врожденного иммунитета
Известны ПИД с нарушениями в
Дефекты рецепторов и сигнальных компонентов врожденного иммунитета
Известны ПИД с нарушениями в

Типы иммунного ответа
Типы иммунного ответа

Типы иммунного ответа
Типы иммунного ответа

Аутовоспалительные синдромы, связанные с дисфункцией врожденного иммунитета за счет изменений в
Аутовоспалительные синдромы, связанные с дисфункцией врожденного иммунитета за счет изменений в

Дефициты системы комплемента
Редкие формы ПИД (в подавляющем большинстве случаев дефекты системы
Дефициты системы комплемента
Редкие формы ПИД (в подавляющем большинстве случаев дефекты системы

Особенности проявления симптомов ПИД
Особенности проявления симптомов ПИД

Сруктура летальности ПИД
Сруктура летальности ПИД

Вопросы
Вопросы

Тестовые вопросы
Изолированное снижение уровня IgA в сыворотке крови определяют при:
ТКИД.
Транзиторной
Тестовые вопросы
Изолированное снижение уровня IgA в сыворотке крови определяют при:
ТКИД.
Транзиторной

Тестовые вопросы
Транзиторная гипогаммаглобулинемия диагностируется в возрасте:
С 6 месяцев до 3
Тестовые вопросы
Транзиторная гипогаммаглобулинемия диагностируется в возрасте:
С 6 месяцев до 3

Тестовые вопросы
При аутовоспалительных нарушениях имеет место дисфункция врожденного иммунитета за
Тестовые вопросы
При аутовоспалительных нарушениях имеет место дисфункция врожденного иммунитета за

Тестовые вопросы
С наибольшей частотой в популяции встречается:
Кожно-слизистый кандидоз
Транзиторная гипогаммаглобулинемия.
Общая вариабельная
Тестовые вопросы
С наибольшей частотой в популяции встречается:
Кожно-слизистый кандидоз
Транзиторная гипогаммаглобулинемия.
Общая вариабельная

Тестовые вопросы
К врожденным дефектам фагоцитарного звена относятся:
Нарушение хемотаксиса.
Нарушения адгезии.
Нарушение эндоцитоза.
Нарушение
Тестовые вопросы
К врожденным дефектам фагоцитарного звена относятся:
Нарушение хемотаксиса.
Нарушения адгезии.
Нарушение эндоцитоза.
Нарушение


 Подготовка к родам
Подготовка к родам Аппарат движения человека
Аппарат движения человека Арт-терапия. Мандалы для детей
Арт-терапия. Мандалы для детей Настойки, в состав которых входит этанол
Настойки, в состав которых входит этанол Сектор по вопросам пропаганды здорового образа жизни в ГБПОУ Самарский медицинский колледж им. Н. Ляпиной
Сектор по вопросам пропаганды здорового образа жизни в ГБПОУ Самарский медицинский колледж им. Н. Ляпиной Система здравоохранения США
Система здравоохранения США Практически нормальное зрение
Практически нормальное зрение Стадії відновлення верхньої кінцівки в пацієнта з ураженням головного мозку
Стадії відновлення верхньої кінцівки в пацієнта з ураженням головного мозку Жыныс жолдары арқылы жұғатын аурулар
Жыныс жолдары арқылы жұғатын аурулар Биомониторинг как этап системы реабилитации. Задачи на 2016 год
Биомониторинг как этап системы реабилитации. Задачи на 2016 год Регионарный кровоток
Регионарный кровоток GLPS
GLPS Экстракты как лекарственная форма. Лекция №5
Экстракты как лекарственная форма. Лекция №5 Патология, синдромология и нозология экзогенно-органического регистра. Органические психозы. Посткоммоционные расстройства
Патология, синдромология и нозология экзогенно-органического регистра. Органические психозы. Посткоммоционные расстройства Узловатая эритема
Узловатая эритема Основи законодавства, нормативні документи та Накази МОЗ України щодо організації лікувально-профілактичної допомоги
Основи законодавства, нормативні документи та Накази МОЗ України щодо організації лікувально-профілактичної допомоги Выбор друзей и компании
Выбор друзей и компании Lobular pneumonia
Lobular pneumonia Магнитно-резонансная томография
Магнитно-резонансная томография Status lokalis. Пародонт
Status lokalis. Пародонт Очаговый туберкулез. Инфильтративный туберкулез. Казеозная пневмония. Туберкулема
Очаговый туберкулез. Инфильтративный туберкулез. Казеозная пневмония. Туберкулема Бірыңғай ұлттық денсаулық сақтау жүйесі
Бірыңғай ұлттық денсаулық сақтау жүйесі Основы иммуногематологии. Значение антигенов в трансфузиологии, акушерстве
Основы иммуногематологии. Значение антигенов в трансфузиологии, акушерстве Клинические и параклинические методы обследования
Клинические и параклинические методы обследования Форменные элементы крови. Группы крови
Форменные элементы крови. Группы крови Жалобы больных при заболеваниях сердечнососудистой системы
Жалобы больных при заболеваниях сердечнососудистой системы Физиология питания
Физиология питания Физикальное исследование сердечно-сосудистой системы. Часть I
Физикальное исследование сердечно-сосудистой системы. Часть I