- Наследственные иммунодефициты

Содержание
- 2. Наследственные иммунодефицитные состояния — это гетерогенная группа болезней, характеризующаяся недостаточностью иммунной системы вследствие генетических дефектов, приводящих
- 3. Клинические проявления иммунодефицитных состояний обусловлены снижением активности или неспособностью организма к эффективному осуществлению реакций клеточного и/или
- 4. Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1 : 25000 до 1 : 50000.
- 5. Недостаточность системы В-лимфоцитов и гуморального звена иммунитета выявляется у 50-75% из общего числа больных с различными
- 6. В зависимости от преимущественного повреждения клеток иммунной системы все иммунодефициты разделены на 4 группы
- 7. (1) Т-зависимые, клеточные с повреждением клеточного иммунитета, (2) В-зависимые, гуморальные с повреждением гуморального иммунитета, (3) А-зависимые
- 8. Важнейшая роль в производстве антител принадлежит генам иммуноглобулинов и Т-клеточных рецепторов, синтезируемых соответственно В- и Т-лимфоцитами
- 9. В ходе созревания лимфоцитов от стволовой клетки до развития их специализированных субпопуляций происходит уникальная перестройка этих
- 10. Цитокиновая система обеспечивает взаимодействие между лимфоцитами и фагоцитами, координируя тем самым работу разнообразных клеток, задействованных в
- 11. В основе развития наследственных иммунодефицитов могут лежать многие процессы, затрагивающие, в частности, (1) созревание лимфоцитов, фагоцитов
- 12. При наиболее тяжелых формах комбинированного иммунодефицита нарушается процесс дифференцировки Т-лимфоцитов. Нарушения дифференцировки В-лимфоцитов приводят к простому
- 13. При хроническом гранулематозе наблюдается недостаточность фагоцитарной системы иммунитета. Иммунодефицитные состояния входят в состав нескольких десятков моногенных
- 14. Экспрессия генов иммуноглобулинов и Т-клеточных рецепторов
- 15. Организм каждого человека может вырабатывать около 1011 различных антител, и очевидно, что необходимая для этого информация
- 16. Разнообразие генов иммуноглобулинов, контролирующих синтез антител, достигается за счет их физической перегруппировки в соматических клетках и
- 17. Изначально антитела кодируются относительно небольшим количеством генов, но в ходе созревания В-клеток происходит уникальная соматическая перестройка
- 18. Эта перестройка сопровождается резким увеличением частоты соматических мутаций, что, в конечном счете, и приводит к огромному
- 19. Молекулы иммуноглобулинов формируются из двух тяжелых (H) и двух легких (L) цепей, причем каждая из этих
- 20. Постоянный участок определяет класс иммуноглобулинов (M, G, A, E или D), и его аминокислотная последовательность относительно
- 21. Аминокислотная последовательность V-участков очень изменчива у разных антител. V-участки H- и L-цепей определяют специфичность антитела, так
- 22. В половых клетках человека нет полноразмерных генов, кодирующих H- и L-цепи, но существуют множества гомологичных генов,
- 23. Так, локус для V-участка H-цепи состоит из трех сегментов V, D и J. В первом сегменте
- 24. Причем гены вариабельного участка перемежаются с генами константного участка иммуноглобулинов разных типов. Таким образом, общая площадь
- 25. В ходе дифференцировки клеток, продуцирующих антитела, ДНК в локусах иммуноглобулинов перестраивается, создавая функциональные гены для H-
- 26. Так, при образовании полноразмерного готового к транскрипции гена для H-цепи происходит разрыв двойной спирали ДНК с
- 27. Аналогично перестраиваются гены для L-цепи иммуноглобулинов. При неточном соединении сегментов генов в процессе соматической перестройки могут
- 28. При антигенной стимуляции начинают размножаться и подвергаться частым точечным мутациям в пределах перестроенной последовательности ДНК те
- 29. При этом частота спонтанных мутаций оказывается на 2-3 порядка выше, чем где-либо в геноме. Эти мутации,
- 30. Таким образом, разнообразие антител достигается за счет (1) использования двух различных цепей (H и L) иммуноглобулинов,
- 31. Сходный механизм соматической перегруппировки характерен и для семейства генов Т-клеточного рецептора (TCR), продуктом которых является трансмембранный
- 32. Этот белок имеет структурное сходство с иммуноглобулинами. Все его цепи имеют как постоянные, так и вариабельные
- 33. Подчеркнем еще раз, что перестройка генетического материала происходит только в В- и Т-клетках и касается исключительно
- 34. Тяжелый комбинированный иммунодефицит
- 35. Тяжелый комбинированный иммунодефицит (ТКИД) — это гетерогенная группа болезней, обусловленных нарушениями клеточного и гуморального иммунитета
- 36. Начиная с младенческого возраста, эти больные подвержены рекуррентным инфекционным процессам, развивающимся вследствие активации условно-патогенных микроорганизмов, включая
- 37. У больных наблюдается выраженная лимфопения, сопровождающаяся снижением или полным отсутствием иммуноглобулинов. Общей характеристикой всех типов комбинированного
- 38. Продолжительность жизни больных при отсутствии лечения обычно не превышает одного года. Чаще всего, единственная возможность терапии
- 39. ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов – B+ТКИД и B-ТКИД. В
- 40. Наиболее частым генетическим типом ТКИД является Х-сцепленный комбинированный иммунодефицит, обусловленный мутациями в гене гамма-рецептора интерлейкина 2
- 41. В шести интерлейкиновых рецепторных комплексах присутствует общая гамма-цепь, кодируемая геном IL2RG. Все они являются гетеродимерами и
- 42. У больных мальчиков с гемизиготными мутациями в гене IL2RG функция этих комплексов частично или полностью блокирована,
- 43. К такой же форме (T-, B+, НК-) относится аутосомно-рецессивный B+ТКИД, обусловленный мутациями в гене JAK3. Продукт
- 44. Форма (T-, B+, NK+) ТКИД также генетически гетерогенна и может быть вызвана дефектами цитокиновых рецепторов (IL7R),
- 45. У 20-30% больных комбинированным иммунодефицитом присутствуют клетки-киллеры, при этом отсутствуют Т- и В-лимфоциты – форма (T-,
- 46. Более чем у половины таких больных обнаруживаются мутации в одном из двух соседних синхронно регулируемых генов
- 47. Продукты этих генов являются активаторами рекомбиназы, необходимой для перестройки V-, D- и J-сегментов, которая происходит при
- 48. Многие ферменты репарации ДНК одновременно участвуют в V(D)J-рекомбинации. Этот процесс начинается с образования двунитевого разрыва, который,
- 49. Для каждого из этих шагов необходимы продукты генов RAG1 и RAG2. Различные мутации в этих генах
- 50. Формы (T-, B-, NK+), сопровождающиеся повышенной чувствительностью к ионизирующему облучению, могут быть обусловлены мутациями в одном
- 51. Продукты генов DCLRE1C и NHEJ участвуют в репарации ДНК и V(D)J-рекомбинации на стадиях образования шпилечной структуры
- 52. (T-, B-, NK-)-форма комбинированного иммунодефицита, или наследственная недостаточность аденозиндезаминазы (АДА) клинически является наиболее тяжелой и сопровождается
- 53. Это редкое аутосомно-рецессивное заболевание, встречающееся с частотой 1:100 000. АДА необходима для расщепления пуринов. При ее
- 54. АДА-недостаточность составляет около 15% всех случаев ТКИД и около трети – его аутосомно-рецессивных форм. Клинический полиморфизм
- 55. Наиболее часто болезнь проявляется уже в младенческом возрасте и может быстро приводить к летальному исходу. У
- 56. При асимптомных формах АДА-недостаточности активность фермента в ядерных клетках крови может составлять от 5 до 80%
- 57. Первое успешное лечение АДА-недостаточности методами генотерапии было осуществлено более 20 лет назад в США. Девочке 4-х
- 58. Лечебный эффект длился несколько месяцев, после чего процедуру повторяли с интервалом в 3-5 месяцев. На протяжении
- 59. В результате лечения состояние пациентки настолько улучшилось, что она смогла вести нормальный образ жизни. Столь же
- 60. В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что генетическая конструкция, содержащая нормальный ген
- 61. При этих условиях эффект от каждой процедуры реинфузии пролонгируется. После 2-3 лет подобной терапии пациенты, как
- 62. К числу тяжелых комбинированных иммунодефицитов относится синдром «голых» лимфоцитов (СГЛ), при котором на поверхности антигенпрезентирующих клеток
- 63. При этом становится невозможным Т-зависимый иммунный ответ, хотя количество Т- и В-лимфоцитов в крови нормальное. Заболевание
- 64. Первыми симптомами СГЛ являются упорная диарея, кандидоз кожи и слизистых, интерстициальная пневмония и различные бактериальные инфекции
- 65. В настоящее время идентифицированы гены при двух типах СГЛ – I и II. Более распространенным является
- 66. В настоящее время выделяют 5 групп комплементации СГЛ II типа, обусловленных мутациями в генах различных регуляторных
- 67. При редком СГЛ I типа на поверхности лимфоцитов отсутствуют молекулы класса I HLA, в то время
- 68. Характерными клиническими проявлениями являются бронхоэктазы, эмфизема, панбронхиолиты, бронхиальная обструкция, носовые полипы и синуситы
- 69. СГЛ I типа также представляет собой гетерогенную группу заболеваний, обусловленных мутациями в трех тандемно расположенных в
- 70. Продуктами этих генов являются АТФ-связывающие транспортные белки, участвующие в транслокации необходимых цитоплазматических пептидов к ожидающим их
- 71. Простой вариабельный иммунодефицит
- 72. Простой вариабельный иммунодефицит (ПВИД) связан с нарушением передачи сигналов от Т- к В-клеткам. Ведущим проявлением заболевания
- 73. При этом у больных снижена секреция иммуноглобулинов IgG, IgA и в половине случаев – IgM, а
- 74. Клинически болезнь проявляется частыми бактериальными респираторными и кишечными инфекциями, которые могут развиваться в любом возрасте (с
- 75. Это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в генах сигнальных молекул, локализованных на поверхности Т- или
- 76. Это гены поверхностного Т-клеточного рецептора, выполняющего функции индуцибельного ко-стимулятора Т-клеток (ICOS); специфических В-клеточных рецепторов, участвующих в
- 77. А также специфических поверхностных В-клеточных антигенов, входящих в состав В-клеточного ко-рецепторного комплекса, главной функцией которого является
- 78. Хронический гранулематоз
- 79. Хронический гранулематоз (ХГ) обусловлен неспособностью нейтрофилов и моноцитов лизировать поглощённые микроорганизмы с каталазоположительными свойствами, т.е. вырабатывающие
- 80. Главная причина дефектной функции фагоцитов заключается в нарушении окислительного метаболизма в связи с наличием блока в
- 81. В основе патогенеза этого генетически гетерогенного заболевания лежат дефекты структурных или регуляторных субъединиц белков фагоцитарного НАДФ-оксидазного
- 82. Эти дефекты приводят к снижению способности мононуклеарных клеток выступать в качестве антигенпрезентирующих единиц за счет нарушения
- 83. Заболевание обычно проявляется в первые два года жизни ребенка и у девочек протекает легче. Характерным является
- 85. В последующем воспалительные гранулемы и абсцессы возникают в различных органах (чаще всего в легких), при этом
- 86. Появление гранулем связано с неспособностью фагоцитов к разрушению и перевариванию поглощенных микроорганизмов. Частыми инфекционными осложнениями являются
- 87. Диагноз хронического гранулематоза подтверждается при выявлении низкой активности ферментов гексозомонофосфатного шунта и дефекта продукции перекисных соединений
- 88. Больные постоянно нуждаются в антибактериальной терапии, требующейся даже в периоде ремиссии. В зависимости от степени восприимчивости
- 89. либо (2) назначается чередование антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов, оксихинолонов и др.) в сочетании
- 90. В настоящее время идентифицированы 5 генов, мутации в которых приводят к хроническому гранулематозу. Самым частым является
- 91. Аутосомно-рецессивный цитохром b-негативный тип составляет не более 3% случаев заболевания. Он обусловлен мутациями в гене CYBA,
- 92. Остальные аутосомно-рецессивные типы заболевания являются цитохром b-позитивными и могут быть обусловлены мутациями в генах нейтрофильных цитоплазматических
- 93. 90% аутосомных форм и около 30% всех случаев заболевания обусловлены мутациями в гене нейтрофильного цитоплазматического фактора
- 94. 97% мутантных аллелей в гене NCF1 представлены одной делецией 2 нуклеотидов – 75GT. Присутствие мажорной мутации
- 95. Идентифицирован ген при клинически сходном синдроме нейтрофильного иммунодефицита – RAC2. Его продуктом также является один из
- 96. Инфантильный летальный агранулоцитоз , или болезнь Костмана – заболевание с аутосомно-рецессивным типом наследования, обусловленное мутациями в
- 97. Клинические проявления характеризуются рецидивирующими бактериальными инфекциями кожи в области волосистой части головы, перианальной области, слизистых оболочек,
- 98. Вместе с тем, у больных отмечается резистентность к вирусным и грибковым инфекциям. При лабораторном исследовании количество
- 99. Синдромальные формы наследственного иммунодефицита
- 100. В некоторых случаях наследственный иммунодефицит может сочетаться с патологией других систем
- 101. Синдром рекуррентных инфекций, обусловленный гиперпродукцией IgE (гипер-IgE-синдром), или синдром Джоба является первичным иммунодефицитом, проявляющимся хронической экземой,
- 102. Больные имеют характерные грубые черты лица, аномальный прикус в сочетании с гиперрастяжимостью кожи. Этот синдром также
- 103. По аутосомно-рецессивному типу наследуются типы, обусловленные мутациями в двух генах – DOCK8 и CD40. Ген DOCK8
- 104. Аутосомно-доминантный гипер-IgE-синдром, характеризующийся дополнительно комплексом специфических соединительнотканных и скелетных нарушений, обусловлен мутациями в гене STAT3. Продуктом
- 105. При Х-сцепленном лимфопролиферативномй синдроме, или болезни Дункана, наблюдаются тяжелые нарушения регуляции иммунной системы, часто возникающие после
- 106. У больных, перенёсших инфекционный мононуклеоз, нередко развиваются длительное лихорадочное состояние, лимфаденопатия, лимфоцитоз в периферической крови, гепато-
- 107. В комплексный фенотип болезни Дункана входят тяжелый или фатальный мононуклеоз, приобретенная гипогаммаглобулинемия, гемофагоцитарный лимфогистоцитоз, апластическая анемия,
- 108. Заболевание обусловлено мутациями в гене SH2D1A, кодирующем белок сигнальной активации лимфоцитов (SLAM), участвующий в бинаправленной стимуляции
- 109. Сигнальное SLAM-SLAM-связывание, возникающее при взаимодействии между В-клетками и между В- и Т-клетками, ускоряет экспансию и дифференцировку
- 110. По крайней мере, 6 аллельных Х-сцепленных доминантных иммунодефицитов, которые могут сочетаться с эктодермальной дисплазией, обусловлены мутациями
- 111. Продукт гена IKBKG (IKK-гамма), относящийся к консервативному семейству NEMO-киназ, входит в состав киназного комплекса, принимающего участие
- 112. Этот фактор присутствует во множестве клеток, в которых экспрессируются цитокины, хемокины, факторы роста, молекулы клеточной адгезии
- 113. Несвоевременная активация NFKB ассоциирована с воспалительными процессами при аутоиммунных артритах, бронхиальной астме, септическом шоке, фиброзе легких,
- 114. С другой стороны, полное или частичное ингибирование NFKB связано с процессами апоптоза, нарушением дифференцировки иммунных клеток
- 115. Мутации в гене MYD88 являются причиной развития аутосомно-рецессивного синдрома рекуррентных бактериальных гнойных инфекций, включая пневмококковые инвазии
- 116. Основной функцией адапторного белка MyD88 является индукция активации транскрипционного фактора NFKB. В гене MYD88 идентифицированы две
- 117. Мутации в гене STK4 – стресс-индуцируемой серин/треонинкиназы – ответственны за развитие аутосомно-рецессивного синдрома Т-клеточного иммунодефицита, сочетающегося
- 118. У больных наблюдается прогрессирующая потеря Т-клеток, рекуррентные бактериальные, вирусные и грибковые инфекции, бородавки, абсцессы и аутоиммунные
- 119. Stk4-киназа активируется в ответ на действие факторов роста, многих химических соединений, теплового шока, апоптоз-индуцирующих агентов. Более
- 120. Не исключено, что эта форма синдромального иммунодефицита обусловлена нарушением регуляции эпигенетических процессов в ДНК, опосредуемых химическими
- 121. К эпигенетическим болезням, по-видимому, может быть отнесен аутосомно-рецессивный синдром иммунной недостаточности-центромерной нестабильности-лицевых аномалий – ICF-синдром, так
- 122. Продуктом этого гена является метилтрансфераза 3В – фермент, принимающий участие в одной из главных эпигенетических модификаций
- 123. ICF-синдром характеризуется заметным иммунодефицитом, часто проявляющимся в виде хронических респираторных и желудочно-кишечных инфекций, в сочетании с
- 124. Для большинства пациентов продолжительность жизни снижена из-за рецидивирующих бронхиальных и легочных инфекций
- 126. Скачать презентацию

Наследственные иммунодефицитные состояния — это гетерогенная группа болезней, характеризующаяся недостаточностью иммунной
Наследственные иммунодефицитные состояния — это гетерогенная группа болезней, характеризующаяся недостаточностью иммунной

Клинические проявления иммунодефицитных состояний обусловлены снижением активности или неспособностью организма к
Клинические проявления иммунодефицитных состояний обусловлены снижением активности или неспособностью организма к

Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1 :
Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1 :

Недостаточность системы
В-лимфоцитов и гуморального звена иммунитета выявляется у 50-75% из
Недостаточность системы В-лимфоцитов и гуморального звена иммунитета выявляется у 50-75% из

В зависимости от преимущественного повреждения клеток иммунной системы все иммунодефициты разделены
В зависимости от преимущественного повреждения клеток иммунной системы все иммунодефициты разделены

(1) Т-зависимые, клеточные с повреждением клеточного иммунитета,
(2) В-зависимые, гуморальные с
(1) Т-зависимые, клеточные с повреждением клеточного иммунитета, (2) В-зависимые, гуморальные с

Важнейшая роль в производстве антител принадлежит генам иммуноглобулинов и
Т-клеточных рецепторов,
Важнейшая роль в производстве антител принадлежит генам иммуноглобулинов и Т-клеточных рецепторов,

В ходе созревания лимфоцитов от стволовой клетки до развития их специализированных
В ходе созревания лимфоцитов от стволовой клетки до развития их специализированных

Цитокиновая система обеспечивает взаимодействие между лимфоцитами и фагоцитами, координируя тем самым
Цитокиновая система обеспечивает взаимодействие между лимфоцитами и фагоцитами, координируя тем самым

В основе развития наследственных иммунодефицитов могут лежать многие процессы, затрагивающие,
В основе развития наследственных иммунодефицитов могут лежать многие процессы, затрагивающие,

При наиболее тяжелых формах комбинированного иммунодефицита нарушается процесс дифференцировки
Т-лимфоцитов.
Нарушения
При наиболее тяжелых формах комбинированного иммунодефицита нарушается процесс дифференцировки Т-лимфоцитов. Нарушения

При
хроническом гранулематозе
наблюдается недостаточность фагоцитарной системы иммунитета. Иммунодефицитные состояния входят
При хроническом гранулематозе наблюдается недостаточность фагоцитарной системы иммунитета. Иммунодефицитные состояния входят

Экспрессия генов иммуноглобулинов и Т-клеточных рецепторов
Экспрессия генов иммуноглобулинов и Т-клеточных рецепторов

Организм каждого человека может вырабатывать около 1011 различных антител, и очевидно,
Организм каждого человека может вырабатывать около 1011 различных антител, и очевидно,

Разнообразие генов иммуноглобулинов, контролирующих синтез антител, достигается за счет их физической
Разнообразие генов иммуноглобулинов, контролирующих синтез антител, достигается за счет их физической

Изначально антитела кодируются относительно небольшим количеством генов, но в ходе созревания
Изначально антитела кодируются относительно небольшим количеством генов, но в ходе созревания

Эта перестройка сопровождается резким увеличением частоты соматических мутаций, что, в конечном
Эта перестройка сопровождается резким увеличением частоты соматических мутаций, что, в конечном

Молекулы иммуноглобулинов формируются из двух тяжелых (H) и двух легких (L)
Молекулы иммуноглобулинов формируются из двух тяжелых (H) и двух легких (L)

Постоянный участок определяет класс иммуноглобулинов
(M, G, A, E или D),
Постоянный участок определяет класс иммуноглобулинов (M, G, A, E или D),

Аминокислотная последовательность V-участков очень изменчива у разных антител.
V-участки H- и
Аминокислотная последовательность V-участков очень изменчива у разных антител. V-участки H- и

В половых клетках человека нет полноразмерных генов, кодирующих H- и L-цепи,
В половых клетках человека нет полноразмерных генов, кодирующих H- и L-цепи,

Так, локус для V-участка H-цепи состоит из трех сегментов
V, D
Так, локус для V-участка H-цепи состоит из трех сегментов V, D

Причем гены вариабельного участка перемежаются с генами константного участка иммуноглобулинов разных
Причем гены вариабельного участка перемежаются с генами константного участка иммуноглобулинов разных

В ходе дифференцировки клеток, продуцирующих антитела,
ДНК в локусах иммуноглобулинов перестраивается,
В ходе дифференцировки клеток, продуцирующих антитела, ДНК в локусах иммуноглобулинов перестраивается,

Так, при образовании полноразмерного готового к транскрипции гена для
H-цепи происходит
Так, при образовании полноразмерного готового к транскрипции гена для H-цепи происходит

Аналогично перестраиваются гены для L-цепи иммуноглобулинов.
При неточном соединении сегментов генов
Аналогично перестраиваются гены для L-цепи иммуноглобулинов. При неточном соединении сегментов генов

При антигенной стимуляции начинают размножаться и подвергаться частым точечным мутациям в
При антигенной стимуляции начинают размножаться и подвергаться частым точечным мутациям в

При этом частота спонтанных мутаций оказывается на 2-3 порядка выше, чем
При этом частота спонтанных мутаций оказывается на 2-3 порядка выше, чем

Таким образом, разнообразие антител достигается за счет
(1) использования двух различных
Таким образом, разнообразие антител достигается за счет (1) использования двух различных

Сходный механизм соматической перегруппировки характерен и для семейства генов
Т-клеточного рецептора
Сходный механизм соматической перегруппировки характерен и для семейства генов Т-клеточного рецептора

Этот белок имеет структурное сходство с иммуноглобулинами.
Все его цепи имеют
Этот белок имеет структурное сходство с иммуноглобулинами. Все его цепи имеют

Подчеркнем еще раз, что перестройка генетического материала происходит только в В-
Подчеркнем еще раз, что перестройка генетического материала происходит только в В-

Тяжелый комбинированный иммунодефицит
Тяжелый комбинированный иммунодефицит

Тяжелый комбинированный иммунодефицит (ТКИД)
— это гетерогенная группа болезней, обусловленных нарушениями
Тяжелый комбинированный иммунодефицит (ТКИД) — это гетерогенная группа болезней, обусловленных нарушениями

Начиная с младенческого возраста, эти больные подвержены рекуррентным инфекционным процессам, развивающимся
Начиная с младенческого возраста, эти больные подвержены рекуррентным инфекционным процессам, развивающимся

У больных наблюдается выраженная лимфопения, сопровождающаяся снижением или полным отсутствием иммуноглобулинов.
У больных наблюдается выраженная лимфопения, сопровождающаяся снижением или полным отсутствием иммуноглобулинов.

Продолжительность жизни больных при отсутствии лечения обычно не превышает
одного года.
Продолжительность жизни больных при отсутствии лечения обычно не превышает одного года.

ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов
ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов

Наиболее частым генетическим типом ТКИД является Х-сцепленный комбинированный иммунодефицит, обусловленный мутациями
Наиболее частым генетическим типом ТКИД является Х-сцепленный комбинированный иммунодефицит, обусловленный мутациями

В шести интерлейкиновых рецепторных комплексах присутствует общая гамма-цепь, кодируемая геном IL2RG.
В шести интерлейкиновых рецепторных комплексах присутствует общая гамма-цепь, кодируемая геном IL2RG.

У больных мальчиков с гемизиготными мутациями в гене IL2RG функция этих
У больных мальчиков с гемизиготными мутациями в гене IL2RG функция этих

К такой же форме (T-, B+, НК-) относится аутосомно-рецессивный B+ТКИД, обусловленный
К такой же форме (T-, B+, НК-) относится аутосомно-рецессивный B+ТКИД, обусловленный

Форма (T-, B+, NK+) ТКИД также генетически гетерогенна и может быть
Форма (T-, B+, NK+) ТКИД также генетически гетерогенна и может быть

У 20-30% больных комбинированным иммунодефицитом присутствуют клетки-киллеры, при этом отсутствуют Т-
У 20-30% больных комбинированным иммунодефицитом присутствуют клетки-киллеры, при этом отсутствуют Т-

Более чем у половины таких больных обнаруживаются мутации в одном из
Более чем у половины таких больных обнаруживаются мутации в одном из

Продукты этих генов являются активаторами рекомбиназы, необходимой для перестройки V-, D-
Продукты этих генов являются активаторами рекомбиназы, необходимой для перестройки V-, D-

Многие ферменты репарации ДНК одновременно участвуют в V(D)J-рекомбинации.
Этот процесс начинается
Многие ферменты репарации ДНК одновременно участвуют в V(D)J-рекомбинации. Этот процесс начинается

Для каждого из этих шагов необходимы продукты генов
RAG1 и RAG2.
Для каждого из этих шагов необходимы продукты генов RAG1 и RAG2.

Формы (T-, B-, NK+), сопровождающиеся повышенной чувствительностью к ионизирующему облучению, могут
Формы (T-, B-, NK+), сопровождающиеся повышенной чувствительностью к ионизирующему облучению, могут

Продукты генов DCLRE1C и NHEJ участвуют в репарации ДНК и V(D)J-рекомбинации
Продукты генов DCLRE1C и NHEJ участвуют в репарации ДНК и V(D)J-рекомбинации

(T-, B-, NK-)-форма комбинированного иммунодефицита, или наследственная недостаточность аденозиндезаминазы (АДА) клинически
(T-, B-, NK-)-форма комбинированного иммунодефицита, или наследственная недостаточность аденозиндезаминазы (АДА) клинически

Это редкое аутосомно-рецессивное заболевание, встречающееся с частотой 1:100 000.
АДА необходима
Это редкое аутосомно-рецессивное заболевание, встречающееся с частотой 1:100 000. АДА необходима

АДА-недостаточность составляет около 15% всех случаев ТКИД и около трети –
АДА-недостаточность составляет около 15% всех случаев ТКИД и около трети –

Наиболее часто болезнь проявляется уже в младенческом возрасте и может быстро
Наиболее часто болезнь проявляется уже в младенческом возрасте и может быстро

При асимптомных формах АДА-недостаточности активность фермента в ядерных клетках крови может
При асимптомных формах АДА-недостаточности активность фермента в ядерных клетках крови может

Первое успешное лечение
АДА-недостаточности методами генотерапии было осуществлено более 20 лет
Первое успешное лечение АДА-недостаточности методами генотерапии было осуществлено более 20 лет

Лечебный эффект длился несколько месяцев, после чего процедуру повторяли с интервалом
Лечебный эффект длился несколько месяцев, после чего процедуру повторяли с интервалом

В результате лечения состояние пациентки настолько улучшилось, что она смогла вести
В результате лечения состояние пациентки настолько улучшилось, что она смогла вести

В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что
В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что

При этих условиях эффект от каждой процедуры реинфузии пролонгируется.
После 2-3
При этих условиях эффект от каждой процедуры реинфузии пролонгируется. После 2-3

К числу тяжелых комбинированных иммунодефицитов относится синдром «голых» лимфоцитов (СГЛ), при
К числу тяжелых комбинированных иммунодефицитов относится синдром «голых» лимфоцитов (СГЛ), при

При этом становится невозможным Т-зависимый иммунный ответ, хотя количество Т- и
При этом становится невозможным Т-зависимый иммунный ответ, хотя количество Т- и

Первыми симптомами СГЛ являются упорная диарея, кандидоз кожи и слизистых, интерстициальная
Первыми симптомами СГЛ являются упорная диарея, кандидоз кожи и слизистых, интерстициальная

В настоящее время идентифицированы гены при двух типах СГЛ – I
В настоящее время идентифицированы гены при двух типах СГЛ – I

В настоящее время выделяют 5 групп комплементации СГЛ II типа, обусловленных
В настоящее время выделяют 5 групп комплементации СГЛ II типа, обусловленных

При редком СГЛ I типа на поверхности лимфоцитов отсутствуют молекулы класса
При редком СГЛ I типа на поверхности лимфоцитов отсутствуют молекулы класса

Характерными клиническими проявлениями являются бронхоэктазы, эмфизема, панбронхиолиты, бронхиальная обструкция, носовые полипы
Характерными клиническими проявлениями являются бронхоэктазы, эмфизема, панбронхиолиты, бронхиальная обструкция, носовые полипы

СГЛ I типа также представляет собой гетерогенную группу заболеваний, обусловленных мутациями
СГЛ I типа также представляет собой гетерогенную группу заболеваний, обусловленных мутациями

Продуктами этих генов являются АТФ-связывающие транспортные белки, участвующие в транслокации необходимых
Продуктами этих генов являются АТФ-связывающие транспортные белки, участвующие в транслокации необходимых

Простой вариабельный иммунодефицит
Простой вариабельный иммунодефицит

Простой вариабельный иммунодефицит (ПВИД)
связан с нарушением передачи сигналов от Т-
Простой вариабельный иммунодефицит (ПВИД) связан с нарушением передачи сигналов от Т-

При этом у больных снижена секреция иммуноглобулинов
IgG, IgA и в
При этом у больных снижена секреция иммуноглобулинов IgG, IgA и в

Клинически болезнь проявляется частыми бактериальными респираторными и кишечными инфекциями, которые могут
Клинически болезнь проявляется частыми бактериальными респираторными и кишечными инфекциями, которые могут

Это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в генах сигнальных молекул,
Это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в генах сигнальных молекул,

Это гены поверхностного
Т-клеточного рецептора, выполняющего функции индуцибельного ко-стимулятора Т-клеток (ICOS);
Это гены поверхностного Т-клеточного рецептора, выполняющего функции индуцибельного ко-стимулятора Т-клеток (ICOS);

А также специфических поверхностных
В-клеточных антигенов,
входящих в состав В-клеточного ко-рецепторного
А также специфических поверхностных В-клеточных антигенов, входящих в состав В-клеточного ко-рецепторного

Хронический гранулематоз
Хронический гранулематоз

Хронический гранулематоз (ХГ) обусловлен неспособностью нейтрофилов и моноцитов лизировать поглощённые микроорганизмы
Хронический гранулематоз (ХГ) обусловлен неспособностью нейтрофилов и моноцитов лизировать поглощённые микроорганизмы

Главная причина дефектной функции фагоцитов заключается в нарушении окислительного метаболизма в
Главная причина дефектной функции фагоцитов заключается в нарушении окислительного метаболизма в

В основе патогенеза этого генетически гетерогенного заболевания лежат дефекты структурных или
В основе патогенеза этого генетически гетерогенного заболевания лежат дефекты структурных или

Эти дефекты приводят к снижению способности мононуклеарных клеток выступать в качестве
Эти дефекты приводят к снижению способности мононуклеарных клеток выступать в качестве

Заболевание обычно проявляется в первые два года жизни ребенка и у
Заболевание обычно проявляется в первые два года жизни ребенка и у
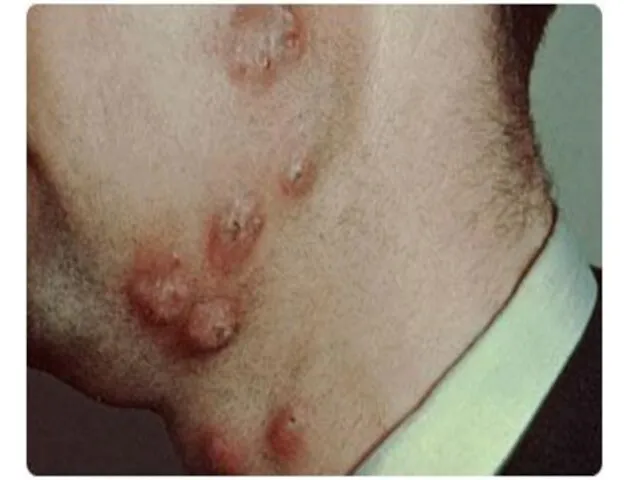

В последующем воспалительные гранулемы и абсцессы возникают в различных органах (чаще
В последующем воспалительные гранулемы и абсцессы возникают в различных органах (чаще

Появление гранулем связано с неспособностью фагоцитов к разрушению и перевариванию поглощенных
Появление гранулем связано с неспособностью фагоцитов к разрушению и перевариванию поглощенных

Диагноз хронического гранулематоза подтверждается при выявлении низкой активности ферментов гексозомонофосфатного шунта
Диагноз хронического гранулематоза подтверждается при выявлении низкой активности ферментов гексозомонофосфатного шунта

Больные постоянно нуждаются в антибактериальной терапии, требующейся даже в периоде ремиссии.
Больные постоянно нуждаются в антибактериальной терапии, требующейся даже в периоде ремиссии.

либо (2) назначается чередование антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов,
либо (2) назначается чередование антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов,

В настоящее время идентифицированы 5 генов, мутации в которых приводят к
В настоящее время идентифицированы 5 генов, мутации в которых приводят к

Аутосомно-рецессивный цитохром b-негативный тип составляет не более 3% случаев заболевания.
Он
Аутосомно-рецессивный цитохром b-негативный тип составляет не более 3% случаев заболевания. Он

Остальные аутосомно-рецессивные типы заболевания являются
цитохром b-позитивными
и могут быть обусловлены
Остальные аутосомно-рецессивные типы заболевания являются цитохром b-позитивными и могут быть обусловлены

90% аутосомных форм и около 30% всех случаев заболевания обусловлены мутациями
90% аутосомных форм и около 30% всех случаев заболевания обусловлены мутациями

97% мутантных аллелей в гене NCF1 представлены одной делецией 2 нуклеотидов
97% мутантных аллелей в гене NCF1 представлены одной делецией 2 нуклеотидов

Идентифицирован ген при клинически сходном
синдроме нейтрофильного иммунодефицита – RAC2.
Его
Идентифицирован ген при клинически сходном синдроме нейтрофильного иммунодефицита – RAC2. Его

Инфантильный летальный агранулоцитоз , или
болезнь Костмана –
заболевание с аутосомно-рецессивным
Инфантильный летальный агранулоцитоз , или болезнь Костмана – заболевание с аутосомно-рецессивным

Клинические проявления характеризуются рецидивирующими бактериальными инфекциями кожи в области волосистой
Клинические проявления характеризуются рецидивирующими бактериальными инфекциями кожи в области волосистой

Вместе с тем, у больных отмечается резистентность к вирусным и грибковым
Вместе с тем, у больных отмечается резистентность к вирусным и грибковым

Синдромальные формы наследственного иммунодефицита
Синдромальные формы наследственного иммунодефицита

В некоторых случаях наследственный иммунодефицит может сочетаться с патологией других систем
В некоторых случаях наследственный иммунодефицит может сочетаться с патологией других систем

Синдром рекуррентных инфекций, обусловленный гиперпродукцией IgE (гипер-IgE-синдром), или
синдром Джоба
является
Синдром рекуррентных инфекций, обусловленный гиперпродукцией IgE (гипер-IgE-синдром), или синдром Джоба является

Больные имеют характерные грубые черты лица, аномальный прикус в сочетании с
Больные имеют характерные грубые черты лица, аномальный прикус в сочетании с

По аутосомно-рецессивному типу наследуются типы, обусловленные мутациями в двух генах –
По аутосомно-рецессивному типу наследуются типы, обусловленные мутациями в двух генах –

Аутосомно-доминантный
гипер-IgE-синдром, характеризующийся дополнительно комплексом специфических соединительнотканных и скелетных нарушений, обусловлен
Аутосомно-доминантный гипер-IgE-синдром, характеризующийся дополнительно комплексом специфических соединительнотканных и скелетных нарушений, обусловлен

При Х-сцепленном лимфопролиферативномй синдроме, или болезни Дункана, наблюдаются тяжелые нарушения регуляции
При Х-сцепленном лимфопролиферативномй синдроме, или болезни Дункана, наблюдаются тяжелые нарушения регуляции

У больных, перенёсших инфекционный мононуклеоз, нередко развиваются длительное лихорадочное состояние, лимфаденопатия,
У больных, перенёсших инфекционный мононуклеоз, нередко развиваются длительное лихорадочное состояние, лимфаденопатия,

В комплексный фенотип болезни Дункана входят тяжелый или фатальный мононуклеоз, приобретенная
В комплексный фенотип болезни Дункана входят тяжелый или фатальный мононуклеоз, приобретенная

Заболевание обусловлено мутациями в гене SH2D1A, кодирующем белок сигнальной активации лимфоцитов
Заболевание обусловлено мутациями в гене SH2D1A, кодирующем белок сигнальной активации лимфоцитов

Сигнальное SLAM-SLAM-связывание, возникающее при взаимодействии между
В-клетками и между
В- и
Сигнальное SLAM-SLAM-связывание, возникающее при взаимодействии между В-клетками и между В- и

По крайней мере, 6 аллельных Х-сцепленных доминантных иммунодефицитов, которые могут сочетаться
По крайней мере, 6 аллельных Х-сцепленных доминантных иммунодефицитов, которые могут сочетаться

Продукт гена IKBKG (IKK-гамма), относящийся к консервативному семейству NEMO-киназ, входит в
Продукт гена IKBKG (IKK-гамма), относящийся к консервативному семейству NEMO-киназ, входит в

Этот фактор присутствует во множестве клеток, в которых экспрессируются цитокины, хемокины,
Этот фактор присутствует во множестве клеток, в которых экспрессируются цитокины, хемокины,

Несвоевременная активация NFKB ассоциирована с
воспалительными процессами
при аутоиммунных артритах,
Несвоевременная активация NFKB ассоциирована с воспалительными процессами при аутоиммунных артритах,

С другой стороны, полное или частичное ингибирование NFKB связано с процессами
С другой стороны, полное или частичное ингибирование NFKB связано с процессами

Мутации в гене MYD88
являются причиной развития аутосомно-рецессивного синдрома рекуррентных бактериальных
Мутации в гене MYD88 являются причиной развития аутосомно-рецессивного синдрома рекуррентных бактериальных

Основной функцией адапторного белка MyD88 является индукция активации транскрипционного фактора NFKB.
Основной функцией адапторного белка MyD88 является индукция активации транскрипционного фактора NFKB.

Мутации в гене STK4 –
стресс-индуцируемой серин/треонинкиназы – ответственны за развитие
Мутации в гене STK4 – стресс-индуцируемой серин/треонинкиназы – ответственны за развитие

У больных наблюдается прогрессирующая потеря
Т-клеток, рекуррентные бактериальные, вирусные и грибковые
У больных наблюдается прогрессирующая потеря Т-клеток, рекуррентные бактериальные, вирусные и грибковые

Stk4-киназа активируется в ответ на действие факторов роста, многих химических соединений,
Stk4-киназа активируется в ответ на действие факторов роста, многих химических соединений,

Не исключено, что эта форма синдромального иммунодефицита обусловлена нарушением регуляции эпигенетических
Не исключено, что эта форма синдромального иммунодефицита обусловлена нарушением регуляции эпигенетических

К эпигенетическим болезням, по-видимому, может быть отнесен аутосомно-рецессивный синдром иммунной недостаточности-центромерной
К эпигенетическим болезням, по-видимому, может быть отнесен аутосомно-рецессивный синдром иммунной недостаточности-центромерной

Продуктом этого гена является метилтрансфераза 3В – фермент, принимающий участие в
Продуктом этого гена является метилтрансфераза 3В – фермент, принимающий участие в

ICF-синдром характеризуется заметным иммунодефицитом,
часто проявляющимся в виде хронических респираторных и
ICF-синдром характеризуется заметным иммунодефицитом, часто проявляющимся в виде хронических респираторных и

Для большинства пациентов продолжительность жизни снижена из-за рецидивирующих бронхиальных и легочных
Для большинства пациентов продолжительность жизни снижена из-за рецидивирующих бронхиальных и легочных
 Осложнения артериальной гипертензии у лиц пожилого и старческого возраста
Осложнения артериальной гипертензии у лиц пожилого и старческого возраста Диагностика и лечение укушенных ран
Диагностика и лечение укушенных ран Клиникалық жағдай. Интоксикационная энцефалопатия головного мозга. Нейропатия лицевого нерва справа
Клиникалық жағдай. Интоксикационная энцефалопатия головного мозга. Нейропатия лицевого нерва справа Вирустық инфекцияларды химиялық препараттармен емдеу
Вирустық инфекцияларды химиялық препараттармен емдеу Хрономедицина и ее разделы
Хрономедицина и ее разделы Антисептика. Виды антисептики
Антисептика. Виды антисептики Дорсопатиялар. Дорсопатия. Жалпы ұғым. Симптомдары және формалары
Дорсопатиялар. Дорсопатия. Жалпы ұғым. Симптомдары және формалары Университет атауы Любляна Медициналык Университеті. Оку процесі
Университет атауы Любляна Медициналык Университеті. Оку процесі Жалпы тәжірибелік дәрігер туралы түсінік
Жалпы тәжірибелік дәрігер туралы түсінік ЭКГ при нарушениях ритма и проводимости
ЭКГ при нарушениях ритма и проводимости Эпителиальные опухоли. Опухоли кроветворной и лимфоидной ткани
Эпителиальные опухоли. Опухоли кроветворной и лимфоидной ткани Кистовидные отёки
Кистовидные отёки Ценности и мотивы дружбы
Ценности и мотивы дружбы Человеческие коммуникации
Человеческие коммуникации Психологическое здоровье детей и укрепление их в условиях УДО
Психологическое здоровье детей и укрепление их в условиях УДО Hyperbarické komory
Hyperbarické komory Профилактика наркозависимости
Профилактика наркозависимости Ведение детей с муковисцидозом в амбулаторных условиях (клинический случай)
Ведение детей с муковисцидозом в амбулаторных условиях (клинический случай) Психотерапияның негізгі әдістері
Психотерапияның негізгі әдістері Наследственные болезни человека
Наследственные болезни человека Общее учение о грыжах (осложненное течение)
Общее учение о грыжах (осложненное течение) Managementul AE Form la contact direct
Managementul AE Form la contact direct Медицина в России XVIII век
Медицина в России XVIII век Роль внеязыковых факторов при передаче мысли словом
Роль внеязыковых факторов при передаче мысли словом Эндокринология. Сахарный диабет. (Лекция 2)
Эндокринология. Сахарный диабет. (Лекция 2) Психофизиологические основы адаптации и дезадаптации к учебной деятельности
Психофизиологические основы адаптации и дезадаптации к учебной деятельности Адам организміне есірткі әсерінің зияндылығын анализдеу және нашақорлықтың алдын - алу
Адам организміне есірткі әсерінің зияндылығын анализдеу және нашақорлықтың алдын - алу Лекция по ветеринарной хирургии
Лекция по ветеринарной хирургии