- Биоинформатическая обработка NGS-данных

Содержание
- 2. Center for Research Informatics, The University of Chicago, Chicago, IL, USA Контроль качества Предварительная обработка Выравнивание
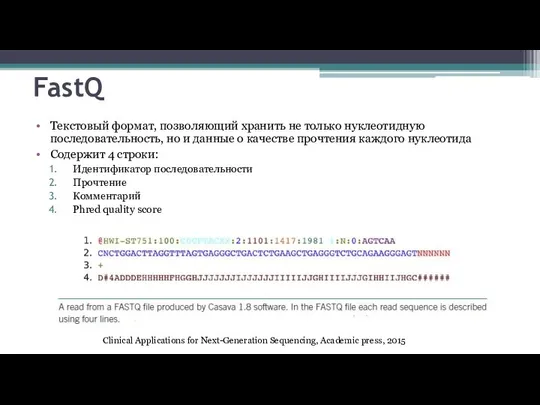
- 3. FastQ Текстовый формат, позволяющий хранить не только нуклеотидную последовательность, но и данные о качестве прочтения каждого
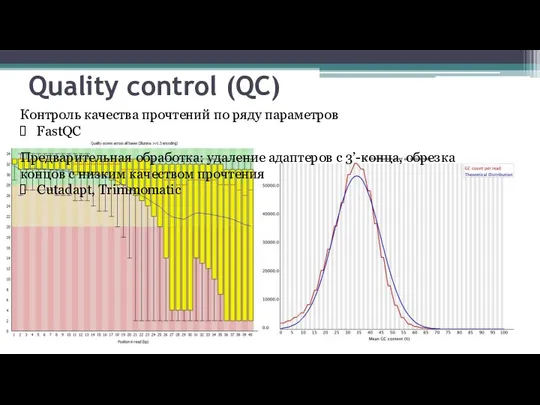
- 4. Quality control (QC) Предварительная обработка: удаление адаптеров с 3’-конца, обрезка концов с низким качеством прочтения Cutadapt,
- 5. Выравнивание (alignment ) AAC - GCTAACGGTAA AACCGCGAAC - - TAA AACGCTAACGGTAA AACCGCGAACTAA BWA, Bowtie2, Novoalign На
- 6. Определение вариантов (variant calling) На этом этапе программа определяет варианты, отличающиеся от референсной последовательности (SNPs, SNVs,
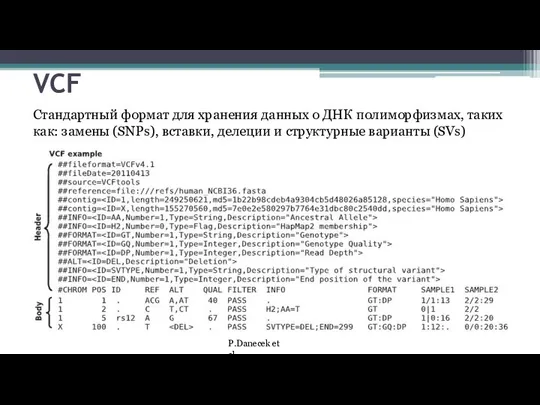
- 7. VCF Стандартный формат для хранения данных о ДНК полиморфизмах, таких как: замены (SNPs), вставки, делеции и
- 8. Аннотация, фильтрация, приоритизация Проводится аннотирование вариантов и предсказание их влияния на кодируемый белок на основе анализа
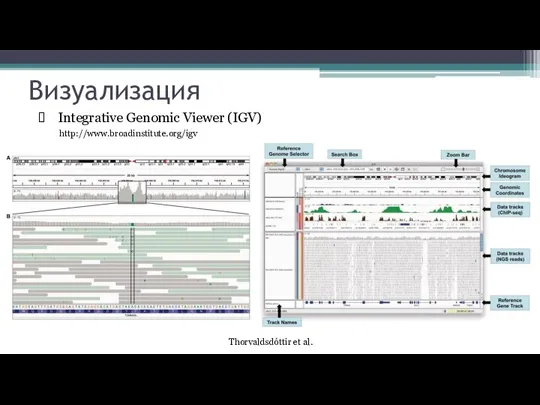
- 9. Визуализация Integrative Genomic Viewer (IGV) http://www.broadinstitute.org/igv Thorvaldsdóttir et al.
- 10. Типы мутаций Nonsense Однонуклеотидные замены, приводящие к возникновению преждевременного терминирующего кодона Мутации с заменой нуклеотида Missense
- 11. Мутации вызванные инсерцией или делецией одного или нескольких нуклеотидов Нормальная ДНК GAA-TGA-CTG-TCT-GGA Нормальный белок Лей-Тре-Асп-Арг-Про Мутантная
- 12. Базы данных геномных вариантов человека Медицинская генетика 2017, №7. Руководство по интерпретации данных, полученных методами массового
- 13. Программы предсказания патогенности вариантов нуклеотидной последовательности (In silico) Медицинская генетика 2017, №7. Руководство по интерпретации данных,
- 14. MutationTaster www.mutationtaster.org Polyphen2 http://genetics.bwh.harvard.edu/pph2/
- 15. Критерии для интерпретации вариантов Для каждого варианта нуклеотидной последовательности специалист подбирает подходящие признаки, которые затем объединяет
- 16. Правила комбинирования критериев для интерпретации вариантов Медицинская генетика 2017, №7. Руководство по интерпретации данных, полученных методами
- 17. Пример медицинского заключения Медицинская генетика 2017, №7. Руководство по интерпретации данных, полученных методами массового параллельного секвенирования
- 19. Скачать презентацию

Center for Research Informatics, The University of Chicago, Chicago, IL, USA
Контроль
Center for Research Informatics, The University of Chicago, Chicago, IL, USA
Контроль
FastQ
Текстовый формат, позволяющий хранить не только нуклеотидную последовательность, но и данные
FastQ
Текстовый формат, позволяющий хранить не только нуклеотидную последовательность, но и данные
Quality control (QC)
Предварительная обработка: удаление адаптеров с 3’-конца, обрезка концов с
Quality control (QC)
Предварительная обработка: удаление адаптеров с 3’-конца, обрезка концов с

Выравнивание (alignment )
AAC - GCTAACGGTAA
AACCGCGAAC - - TAA
AACGCTAACGGTAA
AACCGCGAACTAA
BWA, Bowtie2, Novoalign
На
Выравнивание (alignment )
AAC - GCTAACGGTAA
AACCGCGAAC - - TAA
AACGCTAACGGTAA
AACCGCGAACTAA
BWA, Bowtie2, Novoalign
На

Определение вариантов (variant calling)
На этом этапе программа определяет варианты, отличающиеся от
Определение вариантов (variant calling)
На этом этапе программа определяет варианты, отличающиеся от
VCF
Стандартный формат для хранения данных о ДНК полиморфизмах, таких как: замены
VCF
Стандартный формат для хранения данных о ДНК полиморфизмах, таких как: замены

Аннотация, фильтрация, приоритизация
Проводится аннотирование вариантов и предсказание их влияния на кодируемый
Аннотация, фильтрация, приоритизация
Проводится аннотирование вариантов и предсказание их влияния на кодируемый
Визуализация
Integrative Genomic Viewer (IGV)
http://www.broadinstitute.org/igv
Thorvaldsdóttir et al.
Визуализация
Integrative Genomic Viewer (IGV)
http://www.broadinstitute.org/igv
Thorvaldsdóttir et al.

Типы мутаций
Nonsense
Однонуклеотидные замены, приводящие к возникновению преждевременного терминирующего кодона
Мутации
Типы мутаций
Nonsense
Однонуклеотидные замены, приводящие к возникновению преждевременного терминирующего кодона
Мутации

Мутации вызванные инсерцией или делецией одного или нескольких нуклеотидов
Нормальная
Мутации вызванные инсерцией или делецией одного или нескольких нуклеотидов
Нормальная

Базы данных геномных вариантов человека
Медицинская генетика 2017, №7. Руководство по интерпретации
Базы данных геномных вариантов человека
Медицинская генетика 2017, №7. Руководство по интерпретации

Программы предсказания патогенности вариантов нуклеотидной последовательности (In silico)
Медицинская генетика 2017, №7.
Программы предсказания патогенности вариантов нуклеотидной последовательности (In silico)
Медицинская генетика 2017, №7.
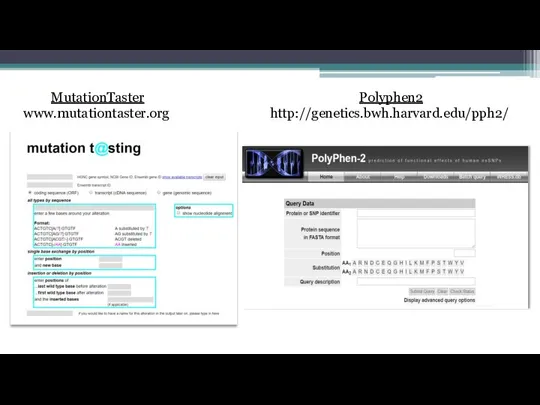
MutationTaster
www.mutationtaster.org
Polyphen2
http://genetics.bwh.harvard.edu/pph2/
MutationTaster
www.mutationtaster.org
Polyphen2
http://genetics.bwh.harvard.edu/pph2/

Критерии для интерпретации вариантов
Для каждого варианта нуклеотидной последовательности специалист подбирает
Критерии для интерпретации вариантов
Для каждого варианта нуклеотидной последовательности специалист подбирает

Правила комбинирования критериев для интерпретации вариантов
Медицинская генетика 2017, №7. Руководство
Правила комбинирования критериев для интерпретации вариантов
Медицинская генетика 2017, №7. Руководство

Пример медицинского заключения
Медицинская генетика 2017, №7. Руководство по интерпретации данных,
полученных
Пример медицинского заключения
Медицинская генетика 2017, №7. Руководство по интерпретации данных,
полученных
 Органы государственной власти РФ
Органы государственной власти РФ Лекція 3. Українська культура пізнього середньовіччя. Друга половина XIII – XVI ст
Лекція 3. Українська культура пізнього середньовіччя. Друга половина XIII – XVI ст Введение в технологии программирования
Введение в технологии программирования Железобетонные фермы
Железобетонные фермы Принципы SOLID. (Лекция 4)
Принципы SOLID. (Лекция 4) Лэтс лёрн хау ту спик фром май харт
Лэтс лёрн хау ту спик фром май харт Решение задач по математике 3 класс
Решение задач по математике 3 класс Архитектура Древнего Египта. Пирамиды
Архитектура Древнего Египта. Пирамиды Презентация на тему "Болезни пищеварения" - скачать презентации по Медицине
Презентация на тему "Болезни пищеварения" - скачать презентации по Медицине Физические свойства. Внешний вид древесины
Физические свойства. Внешний вид древесины Исламский мир. Возникновение ислама
Исламский мир. Возникновение ислама Стихи любимого поэта - презентация для начальной школы_
Стихи любимого поэта - презентация для начальной школы_ Разделение неоднородных систем
Разделение неоднородных систем Гигиена физического воспитания и спортивных сооружений
Гигиена физического воспитания и спортивных сооружений Экономические методы регулирования туристской деятельности
Экономические методы регулирования туристской деятельности Урок алгебры в 7 классе «Линейная функция и её график» Подготовила Татчин У.В. учитель математики МБОУ СОШ №3 город Сургут
Урок алгебры в 7 классе «Линейная функция и её график» Подготовила Татчин У.В. учитель математики МБОУ СОШ №3 город Сургут  Основные понятия стратегического менеджмента
Основные понятия стратегического менеджмента Внеклассное мероприятие «Юный пешеход» Учитель начальных классов МБОУСОШ №44 ст. Северская Краснодарский край Вережникова Е. А.
Внеклассное мероприятие «Юный пешеход» Учитель начальных классов МБОУСОШ №44 ст. Северская Краснодарский край Вережникова Е. А. Общие принципы управления функциями организма
Общие принципы управления функциями организма Лазерная технология 4
Лазерная технология 4 Система управления персоналом
Система управления персоналом Физическое воспитание студентов
Физическое воспитание студентов Единый урок безопасности в сети Интернет
Единый урок безопасности в сети Интернет Розробка бібліотеки для динамічного завантаження виконуваного коду в запущений процес
Розробка бібліотеки для динамічного завантаження виконуваного коду в запущений процес Презентация по МХК Эдуард Мане
Презентация по МХК Эдуард Мане  Динамическое программирование Оптимальные деревья поиска
Динамическое программирование Оптимальные деревья поиска РИСК-МЕНЕЖДМЕНТ Заместитель директора Института менеджмента по науке и дополнительному профессиональному образованию К.
РИСК-МЕНЕЖДМЕНТ Заместитель директора Института менеджмента по науке и дополнительному профессиональному образованию К. Логарифмические неравенства Демонстрационный материал 11 класс
Логарифмические неравенства Демонстрационный материал 11 класс