- Межмолекулярное взаимодействие

Содержание
- 2. Два подхода к квантово-химическому описанию межмолекулярных взаимодействий. 1) Проводится вариационный расчет комплекса АВС… и составляющих его
- 3. Величина суперпозиционной ошибки при расчете энергии межмолекулярного взаимодействия 2) Используется теория возмущений: отдельные молекулы описывают гамильтонианами
- 4. Уравнение Шредингера приобретает вид: (НА+НВ) ΨАnΨВm = (ЕАn+ЕВm) ΨАnΨВm Квантовые числа n и m характеризуют состояния
- 5. Выражение для поляризационной энергии имеет вид Индукционная энергия Еинд отвечает взаимодействию невозмущенной молекулы А с поляризованной
- 6. На больших расстояниях между молекулами R их электронные плотности перекрываются слабо, и каждый из перечисленных вкладов
- 7. Упаковка молекул мочевины в кристалле (штриховые линии - водородные связи). Цифры нумеруют молекулы, расположенные по мере
- 8. Приближенные схемы для больших молекул (биологические и фармакологические задачи) 1. Молекулярный комплекс разбивается на молекулярные фрагменты,
- 9. Оценка ван-дер-ваальсовых атомных радиусов Когда молекулы в кристалле или жидкости связаны лишь ван-дер-ваальсовыми силами, то расстояния
- 10. Донорно-акцепторные молекулярные комплексы Молекулярные комплексы - группы связанных между собой молекул. В них одни из молекул,
- 11. Энергия взаимодействия донора и акцептора ΔЕ геом , ΔЕ эл, ΔЕ обм, ΔЕ поляр, ΔЕ перенос
- 12. Комплексы с переносом заряда например, являются высокопроводящими твердыми органическими материалами. Одно из таких соединений – соль
- 13. Водородная связь Атом Н обладает особой способностью образовывать внутри и между молекулами мостиковые связи X-H∙∙∙Y, соединяясь
- 14. Общий подход к описанию Н-связи не отличается от такового в случае межмолекулярных взаимодействий. Здесь также выделяют
- 15. Сильная Н-связь: небольшая поляризация ЭП неподеленной пары атома О3 к атому Н1 плюс (в результате интерференции
- 16. Специфические невалентные взаимодействия В органических (и металлоорганических кристаллах) кратчайшие расстояния между соседними молекулами меньше, чем сумма
- 17. Структура кристаллического хлора Лапласиан ЭП в кристалле Cl2 (HF 6.21G*)
- 18. Лапласиан ЭП в димере (Cl2)2 (HF 6.311G*)
- 20. Скачать презентацию

Два подхода к квантово-химическому описанию межмолекулярных взаимодействий.
1) Проводится вариационный расчет
Два подхода к квантово-химическому описанию межмолекулярных взаимодействий.
1) Проводится вариационный расчет

Величина суперпозиционной ошибки при расчете энергии межмолекулярного взаимодействия
2) Используется теория
Величина суперпозиционной ошибки при расчете энергии межмолекулярного взаимодействия
2) Используется теория

Уравнение Шредингера приобретает вид:
(НА+НВ) ΨАnΨВm = (ЕАn+ЕВm) ΨАnΨВm
Квантовые числа
Уравнение Шредингера приобретает вид:
(НА+НВ) ΨАnΨВm = (ЕАn+ЕВm) ΨАnΨВm
Квантовые числа

Выражение для поляризационной энергии имеет вид
Индукционная энергия Еинд отвечает взаимодействию невозмущенной
Выражение для поляризационной энергии имеет вид
Индукционная энергия Еинд отвечает взаимодействию невозмущенной

На больших расстояниях между молекулами R их электронные плотности перекрываются слабо,
На больших расстояниях между молекулами R их электронные плотности перекрываются слабо,
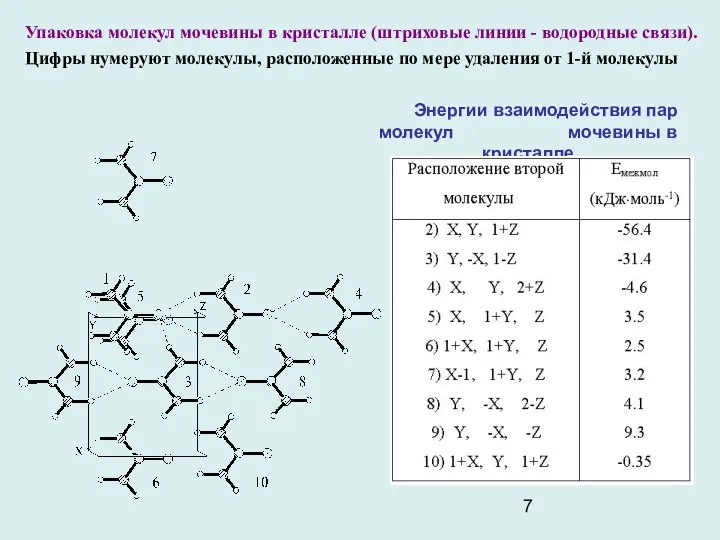
Упаковка молекул мочевины в кристалле (штриховые линии - водородные связи). Цифры
Упаковка молекул мочевины в кристалле (штриховые линии - водородные связи). Цифры

Приближенные схемы для больших молекул (биологические и фармакологические задачи)
1. Молекулярный
Приближенные схемы для больших молекул (биологические и фармакологические задачи)
1. Молекулярный
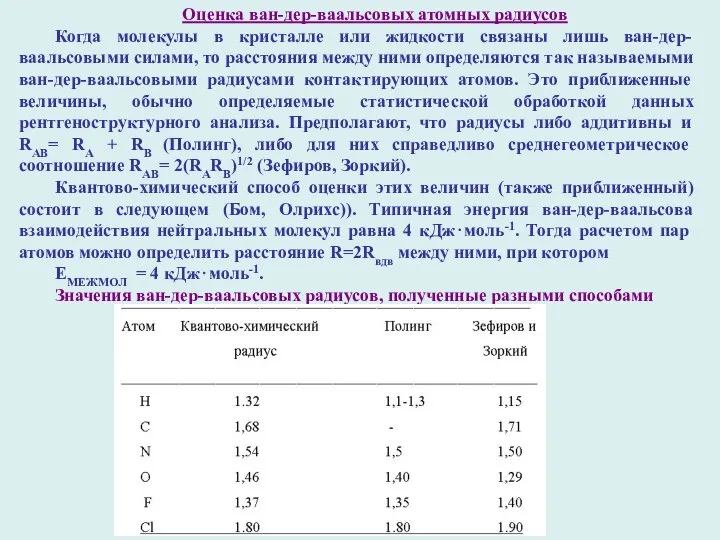
Оценка ван-дер-ваальсовых атомных радиусов
Когда молекулы в кристалле или жидкости связаны лишь
Оценка ван-дер-ваальсовых атомных радиусов
Когда молекулы в кристалле или жидкости связаны лишь

Донорно-акцепторные молекулярные комплексы
Молекулярные комплексы - группы связанных между собой молекул. В
Донорно-акцепторные молекулярные комплексы
Молекулярные комплексы - группы связанных между собой молекул. В

Энергия взаимодействия донора и акцептора
ΔЕ геом , ΔЕ эл, ΔЕ обм,
Энергия взаимодействия донора и акцептора
ΔЕ геом , ΔЕ эл, ΔЕ обм,
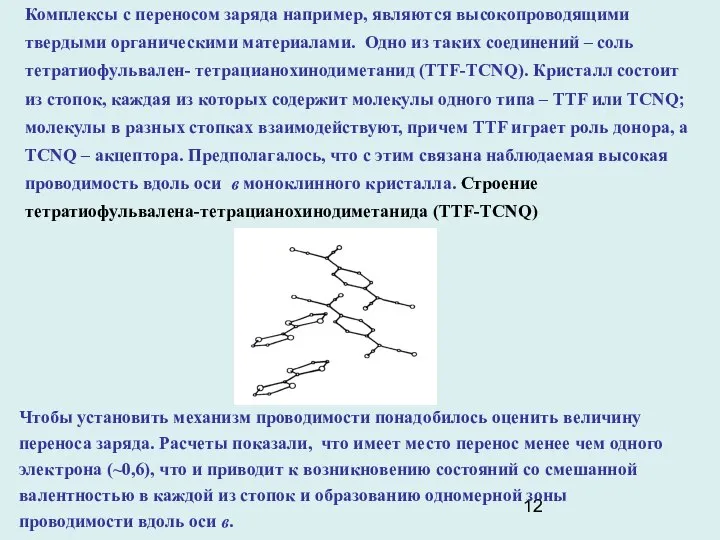
Комплексы с переносом заряда например, являются высокопроводящими твердыми органическими материалами. Одно
Комплексы с переносом заряда например, являются высокопроводящими твердыми органическими материалами. Одно
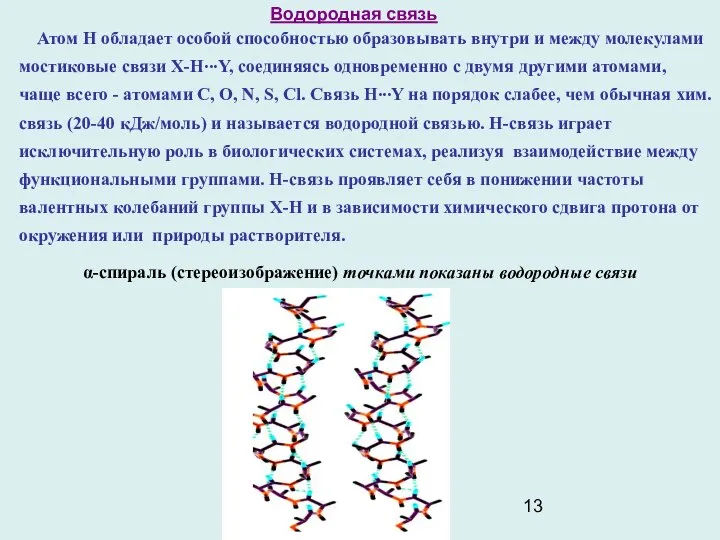
Водородная связь
Атом Н обладает особой способностью образовывать внутри и между
Водородная связь
Атом Н обладает особой способностью образовывать внутри и между

Общий подход к описанию Н-связи не отличается от такового в
Общий подход к описанию Н-связи не отличается от такового в
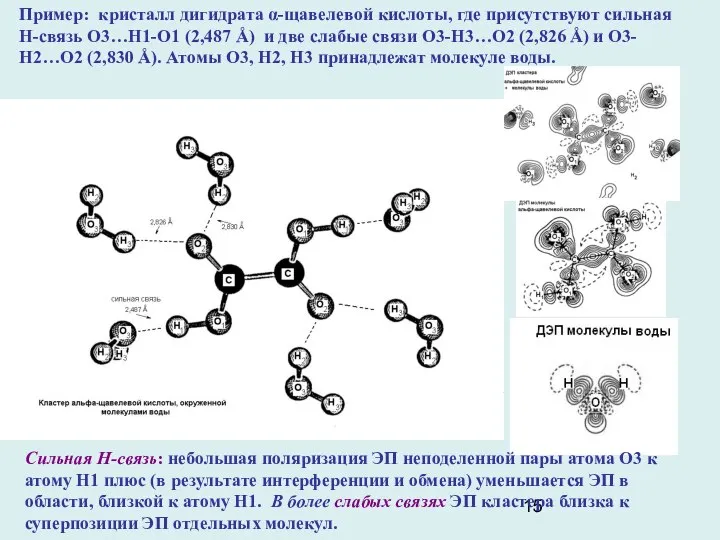
Сильная Н-связь: небольшая поляризация ЭП неподеленной пары атома О3 к атому
Сильная Н-связь: небольшая поляризация ЭП неподеленной пары атома О3 к атому

Специфические невалентные взаимодействия
В органических (и металлоорганических кристаллах) кратчайшие расстояния между
Специфические невалентные взаимодействия
В органических (и металлоорганических кристаллах) кратчайшие расстояния между
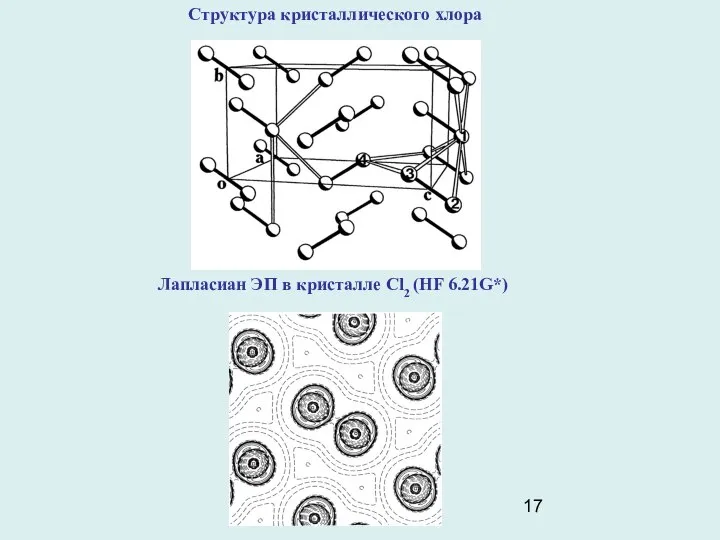
Структура кристаллического хлора
Лапласиан ЭП в кристалле Cl2 (HF 6.21G*)
Структура кристаллического хлора
Лапласиан ЭП в кристалле Cl2 (HF 6.21G*)
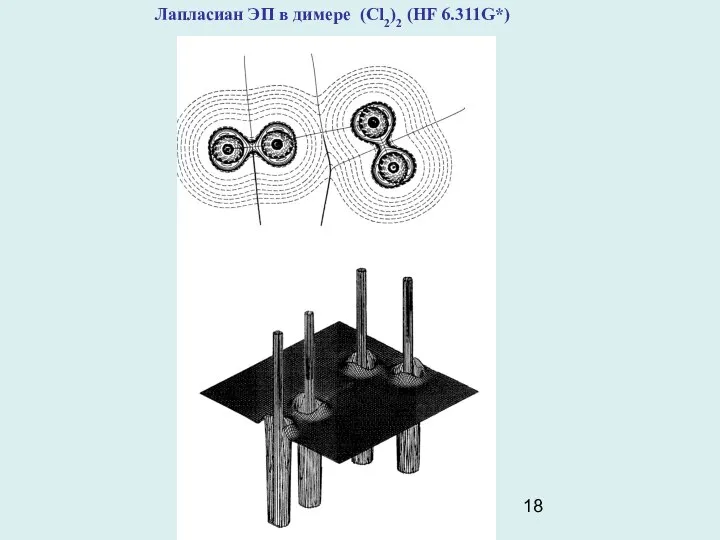
Лапласиан ЭП в димере (Cl2)2 (HF 6.311G*)
Лапласиан ЭП в димере (Cl2)2 (HF 6.311G*)
 Унифицированные системы документации в системе управления персоналом
Унифицированные системы документации в системе управления персоналом Страницы истории Олимпийских игр
Страницы истории Олимпийских игр Информационные технологии в психологии 5
Информационные технологии в психологии 5 Жилой комплекс в Японии, меняющий судьбу
Жилой комплекс в Японии, меняющий судьбу занятие 5 поженились женаты
занятие 5 поженились женаты Паразитологические методы исследования
Паразитологические методы исследования  Основные источники загрязнения окружающей среды и технические меры защиты от загрязнений
Основные источники загрязнения окружающей среды и технические меры защиты от загрязнений Векторы.Линейные операции
Векторы.Линейные операции  Только в боге мы находим силу побеждать (фотографии)
Только в боге мы находим силу побеждать (фотографии) Гражданское право
Гражданское право Организация и техника внешне-экономических операций по купле-продаже лицензий Выполнили: Белоглазова Ю.В., Безнощук Б.Ю.
Организация и техника внешне-экономических операций по купле-продаже лицензий Выполнили: Белоглазова Ю.В., Безнощук Б.Ю.  Ц в е т о в е д е н и е Карточка 4
Ц в е т о в е д е н и е Карточка 4 Презентация на тему "Метод проектів у розвитку якості дошкільної освіти" - скачать презентации по Педагогике
Презентация на тему "Метод проектів у розвитку якості дошкільної освіти" - скачать презентации по Педагогике Хочу быть здоровым! (1 класс) - презентация для начальной школы_
Хочу быть здоровым! (1 класс) - презентация для начальной школы_ Було тихо в Гефсиманії. Н. Н. Німецька мелодія
Було тихо в Гефсиманії. Н. Н. Німецька мелодія Инновационный учебно-методический комплекс "Россия в XIX веке" Визуализированный план-проспект В ИУМК входят: электронный ку
Инновационный учебно-методический комплекс "Россия в XIX веке" Визуализированный план-проспект В ИУМК входят: электронный ку Графики в социально-правовых исследованиях
Графики в социально-правовых исследованиях Объёмное моделирование из гофрированного картона. Стилизованная дымковская игрушка «Индюк»
Объёмное моделирование из гофрированного картона. Стилизованная дымковская игрушка «Индюк» Стратегический выбор. Виды стратегий
Стратегический выбор. Виды стратегий Системы счисления 10 класс
Системы счисления 10 класс  Забойные двигатели: Типы, классификация, устройство. Монтаж и эксплуатация бурового оборудования. Лекция 4
Забойные двигатели: Типы, классификация, устройство. Монтаж и эксплуатация бурового оборудования. Лекция 4 Флаг США
Флаг США Физико-химические основы спекания ультра- и- нанодисперсных порошокв tic- mo, полученных в процессе плазменой переконденсации
Физико-химические основы спекания ультра- и- нанодисперсных порошокв tic- mo, полученных в процессе плазменой переконденсации Регистратор фемтосекундных временных интервалов на основе фотонного эха в тонких пленках
Регистратор фемтосекундных временных интервалов на основе фотонного эха в тонких пленках Тест по простым комбинациям в игре шашки
Тест по простым комбинациям в игре шашки The cultural life of Oxford
The cultural life of Oxford Мат ладьей и королем
Мат ладьей и королем В чем причины изменения литературных направлений начала 19 в.? В чем причины изменения литературных направлений начала 19 в.? Охарак
В чем причины изменения литературных направлений начала 19 в.? В чем причины изменения литературных направлений начала 19 в.? Охарак