- Презентация к уроку английского языка "Food & Drug Administration" - скачать бесплатно

Содержание
- 2. Food & Drug Administration (FDA) 1862, started with a single chemist in the USDA 1927, Bureau
- 3. Adulteration and misbranding of foods & drugs have always been a problem in the U.S. The
- 4. http://www.fda.gov/cder/about/history/Graphics/OilKingLrg.jpg
- 5. http://www.fda.gov/cder/about/history/Gallery/gallery21.htm
- 6. Harvey Washington Wiley, chief chemist concerned about chemical preservatives, initiated "poison squad" experiments Healthy volunteers consumed
- 7. Wiley became convinced that chemical preservatives should be used in food only when necessary That the
- 8. Food and Drugs Act of 1906 First nationwide consumer protection law made it illegal to distribute
- 9. There were however, shortcomings in the 1906 law Law prevented blatant fraud, but it did not
- 10. http://www.fda.gov/oc/history/slideshow/Slide_182_139.html Flavoring Extract Bottle thick glass obscures how much expensive flavoring extract is really in the
- 11. Lash-Lure, an eyelash dye that blinded many women http://www.fda.gov/oc/history/
- 12. A disaster in 1937 prompted Congress to act A Tennessee drug company marketed a form of
- 13. http://www.fda.gov/cder/about/history/Page18.htm Elixir Sulfanilamide
- 14. 1938 Federal Food, Drug, and Cosmetic Act For the first time, required companies to prove the
- 15. Currently the FDA is charged with: Safeguarding the nations food supply, by ensuring that all ingredients
- 16. Medical products need to be proven safe and effective before they can be used by patients
- 17. Drug Review & Approval
- 19. The majority of prospective new drugs fail testing, many never make it passed the pre-clinical stage
- 20. If a compound shows promise during the pre-clinical phase the drug maker may decide to move
- 21. Investigational New Drug Application (IND) The IND is reviewed by both the FDA and a local
- 22. After approval by the IRB and the FDA, clinical trials can begin There are up to
- 23. Phase I Studies Conducted in healthy volunteers, between 20 to 80 Goal is to determine safety
- 24. Phase II Studies Shift in emphasis from safety to effectiveness Collection of preliminary data on whether
- 25. Phase III Studies If effectiveness is shown during phase II the study is expanded to a
- 26. Phase IV Studies Occur after a drug is approved Explore other aspects of the drug such
- 27. New Drug Application (NDA) Once clinical trials are finished the sponsor places a formal request with
- 28. Review Process The review team analyzes all aspects of the study results looking for possible problems,
- 29. If the FDA decides that the benefits of the drug outweigh any risks the drug can
- 30. Accelerated Approval Given to drugs for serious and life-threatening illnesses that lack satisfactory treatments Uses surrogate
- 31. http://www.fda.gov/cder/handbook/develop.htm
- 32. Abbreviated New Drug Application (ANDA) Provides for the review and ultimate approval of a generic drug
- 33. Bioequivalence In order to demonstrate bioequivalence scientists measure the time it takes the generic drug to
- 34. Orphan Drugs In 1982 Congress passed the Orphan Drug Act The goal was to promote the
- 35. Adverse Events Reporting System (AERS) The FDA requires manufacturers to report adverse drug reactions Health care
- 36. If a drug has severe side effects, but is kept on the market a black box
- 37. Recent Drug Controversies Vioxx, voluntarily withdrawn due to increases in heart attacks & strokes Prempro, Premarin;
- 38. If you have questions about medications your or your family are taking: http://www.fda.gov/medwatch/index.html
- 40. Скачать презентацию

Food & Drug Administration (FDA)
1862, started with a single chemist in
Food & Drug Administration (FDA)
1862, started with a single chemist in

Adulteration and misbranding of foods & drugs have always been a
Adulteration and misbranding of foods & drugs have always been a

http://www.fda.gov/cder/about/history/Graphics/OilKingLrg.jpg
http://www.fda.gov/cder/about/history/Graphics/OilKingLrg.jpg

http://www.fda.gov/cder/about/history/Gallery/gallery21.htm
http://www.fda.gov/cder/about/history/Gallery/gallery21.htm

Harvey Washington Wiley, chief chemist concerned about chemical preservatives, initiated "poison
Harvey Washington Wiley, chief chemist concerned about chemical preservatives, initiated "poison

Wiley became convinced that chemical preservatives should be used in food
Wiley became convinced that chemical preservatives should be used in food

Food and Drugs Act of 1906
First nationwide consumer protection law made
Food and Drugs Act of 1906
First nationwide consumer protection law made

There were however, shortcomings in the 1906 law
Law prevented blatant fraud,
There were however, shortcomings in the 1906 law
Law prevented blatant fraud,

http://www.fda.gov/oc/history/slideshow/Slide_182_139.html
Flavoring Extract Bottle
thick glass obscures how much expensive flavoring extract is
http://www.fda.gov/oc/history/slideshow/Slide_182_139.html
Flavoring Extract Bottle
thick glass obscures how much expensive flavoring extract is

Lash-Lure, an eyelash dye that blinded many women
http://www.fda.gov/oc/history/
Lash-Lure, an eyelash dye that blinded many women
http://www.fda.gov/oc/history/

A disaster in 1937 prompted Congress to act
A Tennessee drug company
A disaster in 1937 prompted Congress to act
A Tennessee drug company

http://www.fda.gov/cder/about/history/Page18.htm
Elixir Sulfanilamide
http://www.fda.gov/cder/about/history/Page18.htm
Elixir Sulfanilamide

1938 Federal Food, Drug, and Cosmetic Act
For the first time, required
1938 Federal Food, Drug, and Cosmetic Act
For the first time, required

Currently the FDA is charged with:
Safeguarding the nations food supply, by
Currently the FDA is charged with:
Safeguarding the nations food supply, by

Medical products need to be proven safe and effective before they
Medical products need to be proven safe and effective before they

Drug Review & Approval
Drug Review & Approval
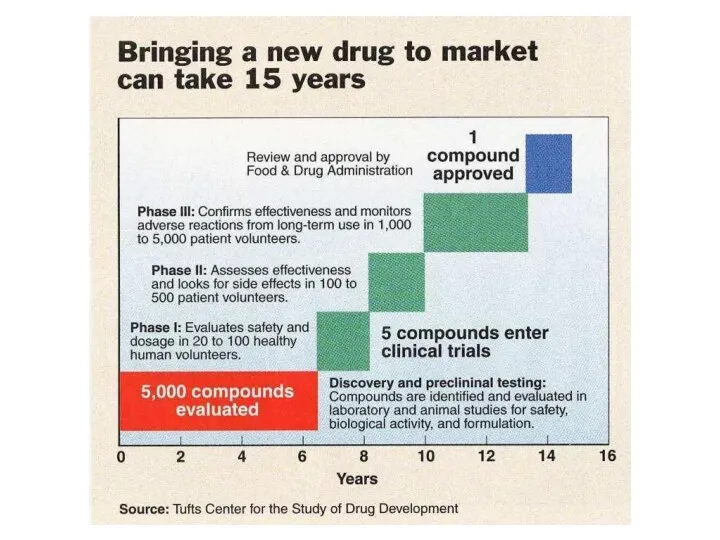

The majority of prospective new drugs fail testing, many never make
The majority of prospective new drugs fail testing, many never make

If a compound shows promise during the pre-clinical phase the drug
If a compound shows promise during the pre-clinical phase the drug

Investigational New Drug Application (IND)
The IND is reviewed by both the
Investigational New Drug Application (IND)
The IND is reviewed by both the

After approval by the IRB and the FDA, clinical trials can
After approval by the IRB and the FDA, clinical trials can

Phase I Studies
Conducted in healthy volunteers, between 20 to 80
Goal is
Phase I Studies
Conducted in healthy volunteers, between 20 to 80
Goal is

Phase II Studies
Shift in emphasis from safety to effectiveness
Collection of preliminary
Phase II Studies
Shift in emphasis from safety to effectiveness
Collection of preliminary

Phase III Studies
If effectiveness is shown during phase II the study
Phase III Studies
If effectiveness is shown during phase II the study

Phase IV Studies
Occur after a drug is approved
Explore other aspects of
Phase IV Studies
Occur after a drug is approved
Explore other aspects of

New Drug Application (NDA)
Once clinical trials are finished the sponsor places
New Drug Application (NDA)
Once clinical trials are finished the sponsor places

Review Process
The review team analyzes all aspects of the study results
Review Process
The review team analyzes all aspects of the study results

If the FDA decides that the benefits of the drug outweigh
If the FDA decides that the benefits of the drug outweigh

Accelerated Approval
Given to drugs for serious and life-threatening illnesses that lack
Accelerated Approval
Given to drugs for serious and life-threatening illnesses that lack
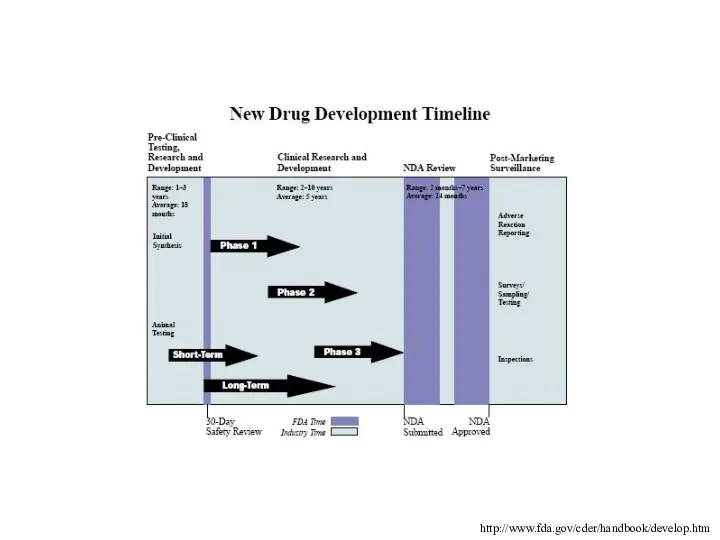
http://www.fda.gov/cder/handbook/develop.htm
http://www.fda.gov/cder/handbook/develop.htm

Abbreviated New Drug Application (ANDA)
Provides for the review and ultimate approval
Abbreviated New Drug Application (ANDA)
Provides for the review and ultimate approval

Bioequivalence
In order to demonstrate bioequivalence scientists measure the time it takes
Bioequivalence
In order to demonstrate bioequivalence scientists measure the time it takes

Orphan Drugs
In 1982 Congress passed the Orphan Drug Act
The goal was
Orphan Drugs
In 1982 Congress passed the Orphan Drug Act
The goal was

Adverse Events Reporting System (AERS)
The FDA requires manufacturers to report adverse
Adverse Events Reporting System (AERS)
The FDA requires manufacturers to report adverse

If a drug has severe side effects, but is kept on
If a drug has severe side effects, but is kept on

Recent Drug Controversies
Vioxx, voluntarily withdrawn due to increases in heart attacks
Recent Drug Controversies
Vioxx, voluntarily withdrawn due to increases in heart attacks

If you have questions about medications your or your family are
If you have questions about medications your or your family are
 Using Modal Verbs
Using Modal Verbs Английский словарь в картинках
Английский словарь в картинках Полиглоты . Работу представила ученица 10 класса Баскаковской МСОШ Айдемирова Саният Учитель Трофимова Е.В.
Полиглоты . Работу представила ученица 10 класса Баскаковской МСОШ Айдемирова Саният Учитель Трофимова Е.В. Fuel normal operation
Fuel normal operation THE WORLD OF ENGLISH LEARNING
THE WORLD OF ENGLISH LEARNING Синтаксис делового письма
Синтаксис делового письма British Museum. Британский музей
British Museum. Британский музей Identify the mistakes please. Present perfect tense and simple past tense
Identify the mistakes please. Present perfect tense and simple past tense Black American english
Black American english Внеурочная деятельность по английскому языку в 1-м классе МОУ Гимназия №4
Внеурочная деятельность по английскому языку в 1-м классе МОУ Гимназия №4  Population Ecology or Demecology
Population Ecology or Demecology Презентация к уроку английского языка "ПРИДАТОЧНЫЕ ПРЕДЛОЖЕНИЯ" - скачать бесплатно
Презентация к уроку английского языка "ПРИДАТОЧНЫЕ ПРЕДЛОЖЕНИЯ" - скачать бесплатно Hello! My name is Alena
Hello! My name is Alena Hometown Выполнила: Ученица 8 класса Башкирова Вероника
Hometown Выполнила: Ученица 8 класса Башкирова Вероника Lesson18
Lesson18 Hairstyles
Hairstyles  Урок по теме: «Биография». Topic: “Biography” Цели и задачи урока: обобщить изученный материал по теме «Биография», повторить и закрепить изученную лексику и грамматику (порядковые числительные), практиковать навыки аудирования, чтения и говорения.
Урок по теме: «Биография». Topic: “Biography” Цели и задачи урока: обобщить изученный материал по теме «Биография», повторить и закрепить изученную лексику и грамматику (порядковые числительные), практиковать навыки аудирования, чтения и говорения. Name of presentation
Name of presentation How to describe people
How to describe people Christmas in New Zealand
Christmas in New Zealand  Тема :путишествие по памятнымместам средневековых государств Европы Выполнил:Пулькин Валера проверила:Вера Валентиновна 2.12
Тема :путишествие по памятнымместам средневековых государств Европы Выполнил:Пулькин Валера проверила:Вера Валентиновна 2.12 Презентация к уроку английского языка "«Немецкий язык. В зоопарке»" - скачать
Презентация к уроку английского языка "«Немецкий язык. В зоопарке»" - скачать  Quiz. I like english. English for children
Quiz. I like english. English for children Do or does? Game
Do or does? Game Королевская семья Великобритании Отношения, семейное древо, тайны и многое другое...
Королевская семья Великобритании Отношения, семейное древо, тайны и многое другое...  Презентация к уроку английского языка "The River Thims" - скачать
Презентация к уроку английского языка "The River Thims" - скачать  Англоязычный политический дискурс: конструирование будущего
Англоязычный политический дискурс: конструирование будущего Halloween lesson plan Form 3-4
Halloween lesson plan Form 3-4