- Катализ органических реакций. (Лекция 15)

Содержание
- 2. Характерно, что небольшие количества Кат ускоряют превращения больших количеств реагирующих веществ. Так на одной части Pt-Кат
- 3. Эффективность Кат часто характеризуют «числом оборотов», а именно числом молей реагентов n, превращенных одним молем Кат
- 4. ОБЩИЕ ЗАКОНОМЕРНОСТИ КАТАЛИЗА Все каталитические процессы – самопроизвольные реакции, протекающие в направлении убыли энергии Гиббса. При
- 5. Энергия активации каталитической реакции значительно меньше, чем той же реакции без катализатора. Например, энергия активации некатализируемой
- 6. Рис. 1. Реакции А→В без Кат. TS0 ΔE0 TS1 TS2 TS3 ΔE1 ΔE2 ΔE3 A B
- 7. Одностадийные процессы катализа (ассоциативные, слитные) протекают по схеме: А + К → TS1 → В +
- 8. Энергии активации обеих стадий меньше, чем в случае реакции без Кат. (ΔЕ2, ΔЕ3 Таким образом, независимо
- 9. ГОМОГЕННЫЙ КАТАЛИЗ Гомогенный катализ можно разделить на: - кислотно-основной; металлокомплексный; окислительно-восстановительный; - гомогенный газофазный; - ферментативный.
- 10. В основном катализе в водных, водно-органических и органических средах в качестве катализаторов используются оксиды и гидроксиды
- 11. Стабилизация алифатических карбкатионов осуществляется за счет индуктивного эффекта и эффекта сверхсопряжения (гиперконьюгации) и увеличивается в ряду
- 12. аллил-катион бензил-катион В случае сопряженных соединений основным фактором, определяющим стабильность карбкатионов, является делокализация положительного заряда за
- 13. Стабильность арилметильных катионов растет в ряду: В этом же ряду уменьшается их реакционная способность. При наличии
- 14. Образование карбкатионов осуществляется следующим образом: 1. Гетеролитическое расщепление σ-связи. Карбкатионы образуются главным образом в полярных растворителях,
- 15. 2. Присоединение электрофила к ненасыщенной функциональной группе. где Протекание реакции зависит от силы электрофила, наличия заместителей,
- 18. При этом в смеси образуется около 50 % наиболее термодинамически стабильного продукта мета-ксилола. Изомеризация ксилолов
- 19. 4. Взаимодействие карбкатионов с нуклеофилами: где X- = F-, Cl-, Br-, CN-, OH--, NH2- где HX
- 20. Карбанионы - это отрицательно заряженные органические ионы с четным числом электронов, отрицательный заряд которых сконцентрирован на
- 21. Алкиланионы представляют собой производные метиланиона, формально генерируемого в результате протолитической реакции. Ввиду малой электроотрицательности углерода для
- 22. Соединения этой группы существенно отличаются по реакционной способности от углеводородных карбанионов. Особую группу карбанионов образуют илиды
- 23. 2. Присоединение нуклеофилов к кратной углерод-углеродной связи Наличие у ненасыщенной связи электроноакцепторных заместителей (—NO2, —ССl3, —CN,
- 24. 2. Замещение у насыщенного атома углерода (SN1, SN2)
- 25. В промышленности многие органические реакции проводится в концентрированных растворах сильных минеральных кислот или оснований в воде,
- 26. Тогда константа кислотности индикатора в растворе будет равна , где и коэффициенты активности. Кислотность среды по
- 27. Имея набор структурно-подобных индикаторов, в качестве которых Гаммет использовал замещенные нитроанилины, можно определить кислотность среды в
- 28. Теория жестких и мягких кислот и оснований Было показано, что легкость протекания кислотно-основной реакции зависит как
- 29. Жесткие основания - основания, в которых атом донора пары электронов имеет высшие занятые орбитали (ВЗМО) с
- 30. При взаимодействии “мягких” кислот с “мягкими” основаниями связь возникает за счет переноса электронов с ВЗМО донора
- 31. СОВРЕМЕННАЯ КЛАССИФИКАЦИЯ КИСЛОТ И ОСНОВАНИЙ Основана на типе орбиталей, принимающих участие в образовании межмолекулярных донорно-акцепторных связей
- 32. Кислотный катализ протонными кислотами Связан с активацией реагента кислотой при протолитическом взаимодействии и последующим превращением активной
- 33. Для насыщенной молекулы, имеющей НПЭ на гетероатоме Х:, при протонировании происходит образование группы (ХН+) с большей
- 34. Для слабых реагентов-оснований, слабых кислот-катализаторов в слабополярных растворителях кислотный катализ идет без полной передачи протона –
- 35. Механизм А-1 заключается в мономолекулярном распаде протонированной молекулы реагента по σ-связи на лимитирующей стадии: (медленно) (быстро)
- 36. По механизму А-1 протекает нитрование некоторых ароматических соединений азотной кислотой, катализируемое серной кислотой. Механизм А-2 включает
- 37. По механизму ААlк2 протекает, например, образование простых эфиров из спиртов: Кислотно-каталитическая реакция карбоновых кислот со спиртами,
- 38. Если в схеме кислотного гидролиза сложных эфиров заменить воду каким-либо спиртом, отличным от образующегося в результате
- 39. Механизм кислотно-каталитического алкилирования бензола олефинами, например, пропиленом можно представить следующей схемой: Отщепление протона в растворе происходит
- 40. Электрофильный катализ или катализ апротонными кислотами Заключается в активации реагента апротонной кислотой – кислотой Льюиса –
- 41. ОСНОВНОЙ КАТАЛИЗ ОК - ускорение реакций в щелочной среде или в присутствии различных оснований. Катализ основаниями,
- 42. По механизму В-2 протекают реакции гидролиза сложных эфиров, галогенангидридов, амидов, этерификация сложных эфиров, альдольная конденсация. Например,
- 43. Нуклеофильный катализ – катализ основаниями, не связанный с кислотно-основным равновесием. Кат – ионы галогенов, оксианионы (OH-,
- 44. В сильно полярных растворителях устанавливается кислотно-основное равновесие: НА + Н2О ↔ Н3+О + А– А– +
- 45. Различают специфический и общий кислотно-основной катализ (КОК). Наблюдается, когда скорость реакции пропорциональна только концентрации иона ОН–
- 46. 0 7 14 pH lgkcat 1 2 3 1 - специфический кислотный катализ; 2 - специфический
- 47. Осуществляется всеми кислотами и основаниями Бренстеда. Для определения этого катализа используют специфичные буферные смеси, где концентрации
- 48. Недостатки гомогенного катализа: необходимость отделения Кат от реакционной массы: образование токсичных сточных вод; повышенный расход Кат
- 51. Разделы для самостоятельной проработки, 2013 Гомогенный металлокомплексный катализ. Лит. 1. Лебедев Н.Н., Манаков М.Н., Швец В.Ф.
- 53. Скачать презентацию

Характерно, что небольшие количества Кат ускоряют превращения больших количеств реагирующих
Характерно, что небольшие количества Кат ускоряют превращения больших количеств реагирующих

Эффективность Кат часто характеризуют «числом оборотов», а именно
Эффективность Кат часто характеризуют «числом оборотов», а именно

ОБЩИЕ ЗАКОНОМЕРНОСТИ КАТАЛИЗА
Все каталитические процессы – самопроизвольные реакции, протекающие в
ОБЩИЕ ЗАКОНОМЕРНОСТИ КАТАЛИЗА
Все каталитические процессы – самопроизвольные реакции, протекающие в

Энергия активации каталитической реакции значительно меньше, чем той же реакции
Энергия активации каталитической реакции значительно меньше, чем той же реакции
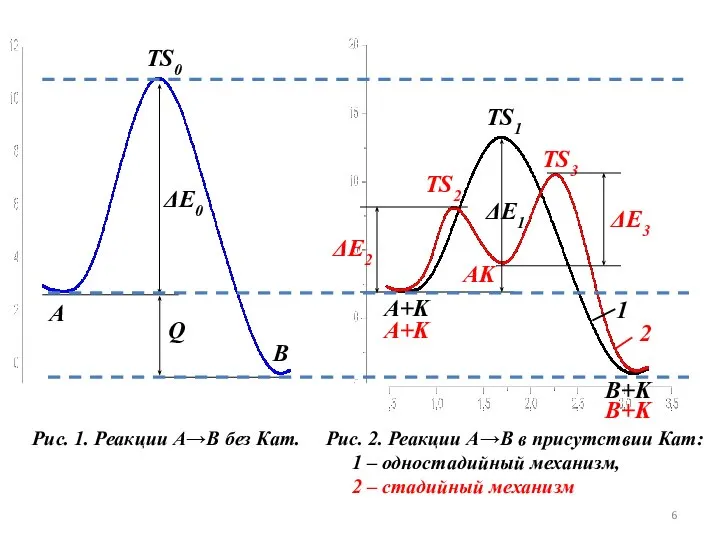
Рис. 1. Реакции А→В без Кат.
TS0
ΔE0
TS1
TS2
TS3
ΔE1
ΔE2
ΔE3
A
B
A+K
Q
B+K
AK
A+K
B+K
Рис. 2. Реакции А→В в
Рис. 1. Реакции А→В без Кат.
TS0
ΔE0
TS1
TS2
TS3
ΔE1
ΔE2
ΔE3
A
B
A+K
Q
B+K
AK
A+K
B+K
Рис. 2. Реакции А→В в

Одностадийные процессы катализа (ассоциативные, слитные) протекают по схеме:
А + К
Одностадийные процессы катализа (ассоциативные, слитные) протекают по схеме:
А + К

Энергии активации обеих стадий меньше, чем в случае реакции без Кат.
(ΔЕ2,
Энергии активации обеих стадий меньше, чем в случае реакции без Кат.
(ΔЕ2,

ГОМОГЕННЫЙ КАТАЛИЗ
Гомогенный катализ можно разделить на:
- кислотно-основной;
металлокомплексный;
окислительно-восстановительный;
- гомогенный газофазный;
-
ГОМОГЕННЫЙ КАТАЛИЗ
Гомогенный катализ можно разделить на:
- кислотно-основной;
металлокомплексный;
окислительно-восстановительный;
- гомогенный газофазный;
-
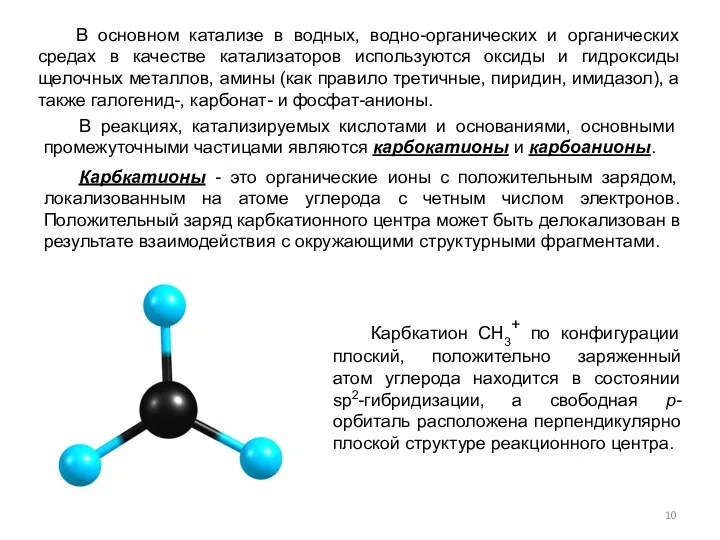
В основном катализе в водных, водно-органических и органических средах в
В основном катализе в водных, водно-органических и органических средах в

Стабилизация алифатических карбкатионов осуществляется за счет индуктивного эффекта и эффекта
Стабилизация алифатических карбкатионов осуществляется за счет индуктивного эффекта и эффекта
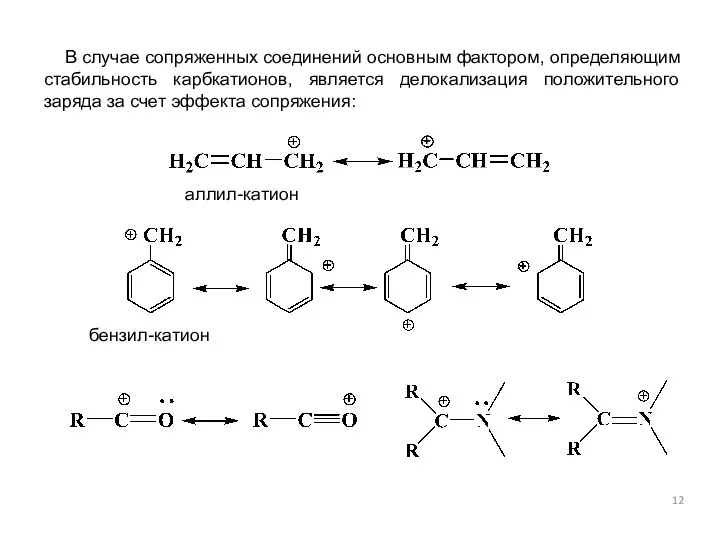
аллил-катион
бензил-катион
В случае сопряженных соединений основным фактором, определяющим стабильность карбкатионов, является
аллил-катион
бензил-катион
В случае сопряженных соединений основным фактором, определяющим стабильность карбкатионов, является

Стабильность арилметильных катионов растет в ряду:
В этом же ряду
Стабильность арилметильных катионов растет в ряду:
В этом же ряду

Образование карбкатионов осуществляется следующим образом:
1. Гетеролитическое расщепление σ-связи.
Карбкатионы
Образование карбкатионов осуществляется следующим образом:
1. Гетеролитическое расщепление σ-связи.
Карбкатионы
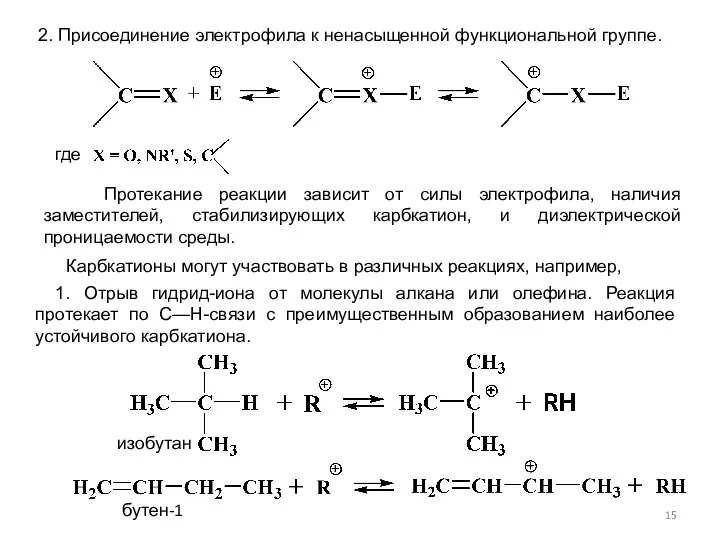
2. Присоединение электрофила к ненасыщенной функциональной группе.
где
Протекание реакции зависит
2. Присоединение электрофила к ненасыщенной функциональной группе.
где
Протекание реакции зависит


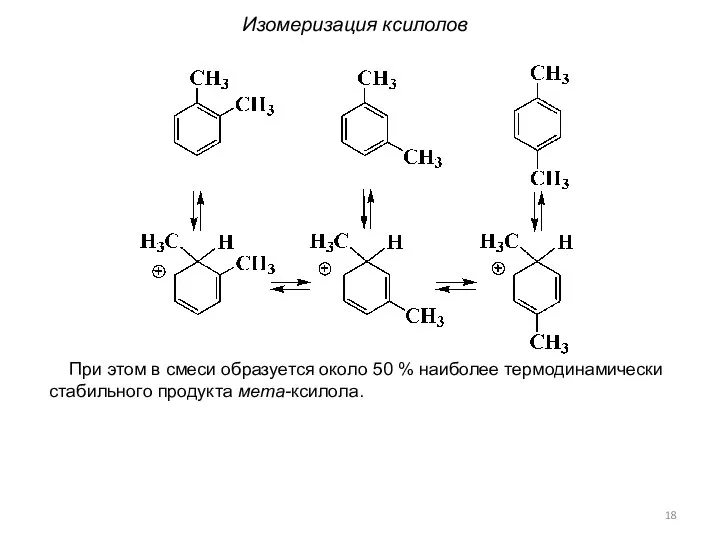
При этом в смеси образуется около 50 % наиболее термодинамически
При этом в смеси образуется около 50 % наиболее термодинамически

4. Взаимодействие карбкатионов с нуклеофилами:
где X- = F-, Cl-, Br-, CN-,
4. Взаимодействие карбкатионов с нуклеофилами:
где X- = F-, Cl-, Br-, CN-,
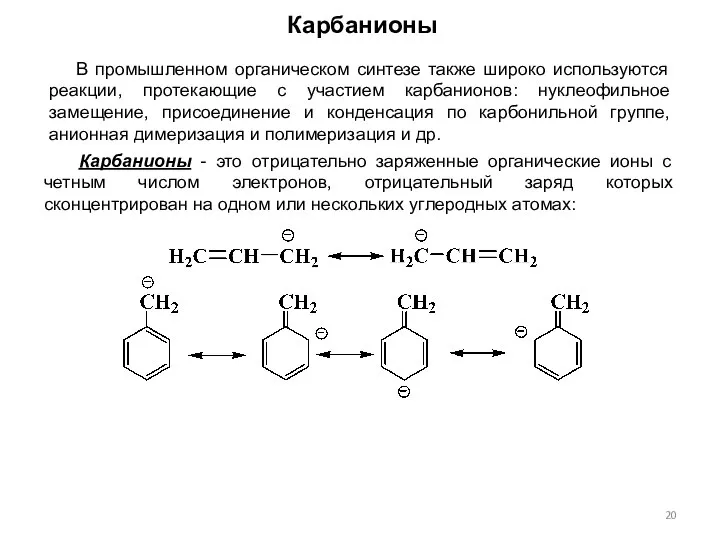
Карбанионы - это отрицательно заряженные органические ионы с четным числом
Карбанионы - это отрицательно заряженные органические ионы с четным числом
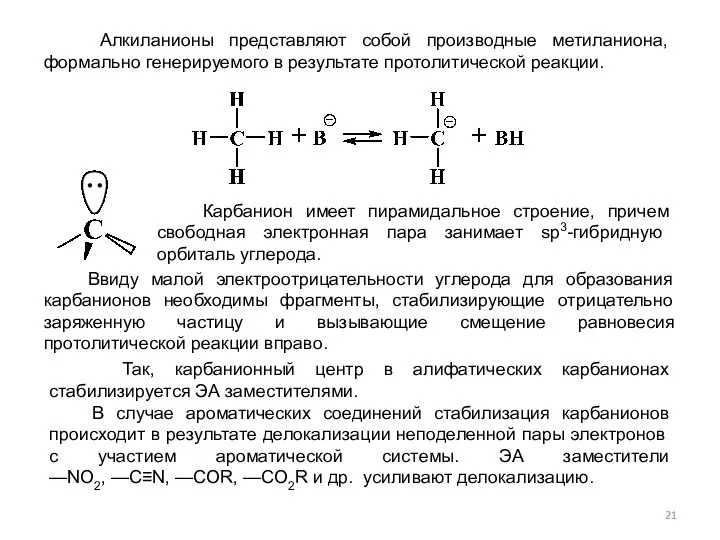
Алкиланионы представляют собой производные метиланиона, формально генерируемого в результате протолитической
Алкиланионы представляют собой производные метиланиона, формально генерируемого в результате протолитической

Соединения этой группы существенно отличаются по реакционной способности от углеводородных
Соединения этой группы существенно отличаются по реакционной способности от углеводородных
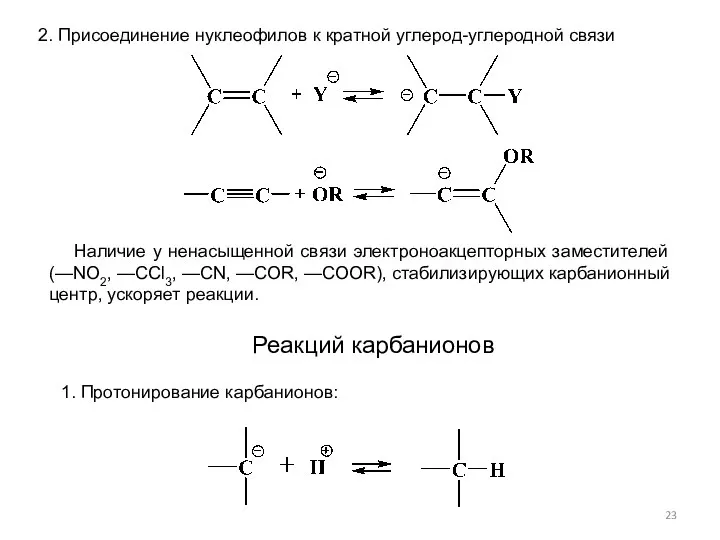
2. Присоединение нуклеофилов к кратной углерод-углеродной связи
Наличие у ненасыщенной связи
2. Присоединение нуклеофилов к кратной углерод-углеродной связи
Наличие у ненасыщенной связи
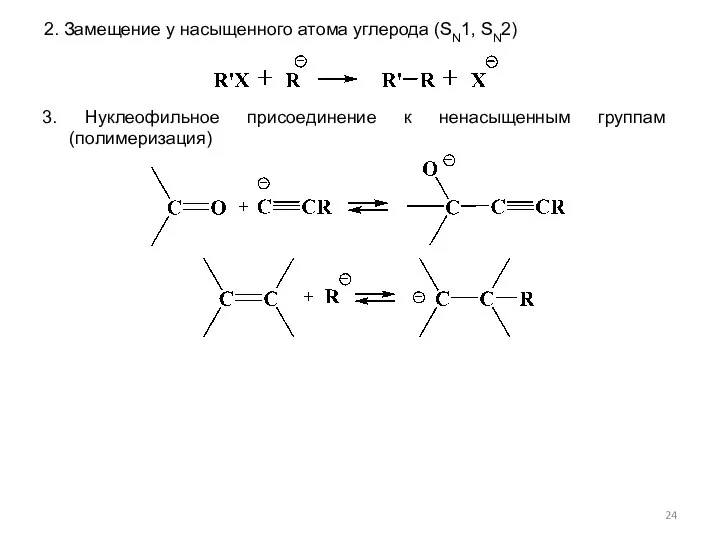
2. Замещение у насыщенного атома углерода (SN1, SN2)
2. Замещение у насыщенного атома углерода (SN1, SN2)

В промышленности многие органические реакции проводится в концентрированных растворах сильных
В промышленности многие органические реакции проводится в концентрированных растворах сильных

Тогда константа кислотности индикатора в растворе будет равна
,
где и коэффициенты активности.
Кислотность
Тогда константа кислотности индикатора в растворе будет равна
,
где и коэффициенты активности.
Кислотность

Имея набор структурно-подобных индикаторов, в качестве которых Гаммет использовал замещенные
Имея набор структурно-подобных индикаторов, в качестве которых Гаммет использовал замещенные

Теория жестких и мягких кислот и оснований
Было показано, что легкость
Теория жестких и мягких кислот и оснований
Было показано, что легкость

Жесткие основания - основания, в которых атом донора пары электронов
Жесткие основания - основания, в которых атом донора пары электронов
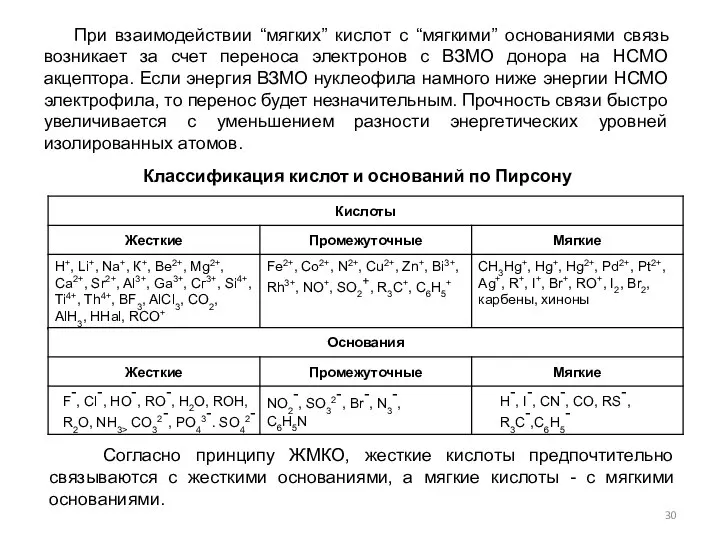
При взаимодействии “мягких” кислот с “мягкими” основаниями связь возникает за
При взаимодействии “мягких” кислот с “мягкими” основаниями связь возникает за

СОВРЕМЕННАЯ КЛАССИФИКАЦИЯ КИСЛОТ И ОСНОВАНИЙ
Основана на типе орбиталей, принимающих участие
СОВРЕМЕННАЯ КЛАССИФИКАЦИЯ КИСЛОТ И ОСНОВАНИЙ
Основана на типе орбиталей, принимающих участие
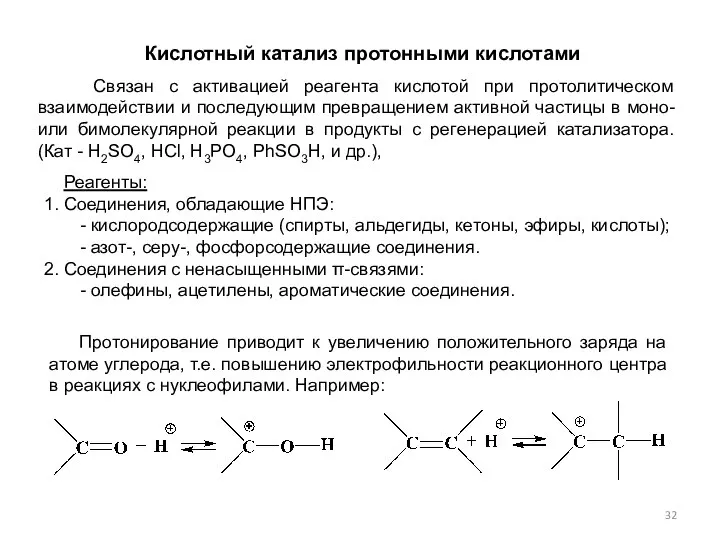
Кислотный катализ протонными кислотами
Связан с активацией реагента кислотой при протолитическом
Кислотный катализ протонными кислотами
Связан с активацией реагента кислотой при протолитическом

Для насыщенной молекулы, имеющей НПЭ на гетероатоме Х:, при протонировании
Для насыщенной молекулы, имеющей НПЭ на гетероатоме Х:, при протонировании
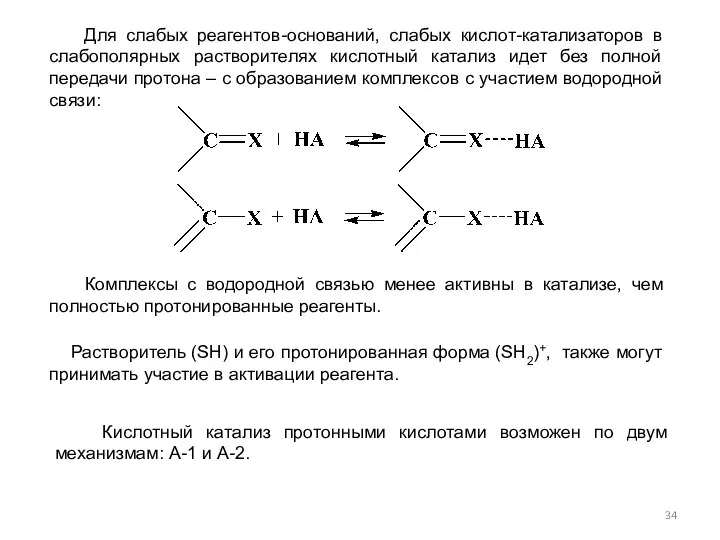
Для слабых реагентов-оснований, слабых кислот-катализаторов в слабополярных растворителях кислотный катализ
Для слабых реагентов-оснований, слабых кислот-катализаторов в слабополярных растворителях кислотный катализ

Механизм А-1 заключается в мономолекулярном распаде протонированной молекулы реагента по
Механизм А-1 заключается в мономолекулярном распаде протонированной молекулы реагента по

По механизму А-1 протекает нитрование некоторых ароматических соединений азотной кислотой,
По механизму А-1 протекает нитрование некоторых ароматических соединений азотной кислотой,
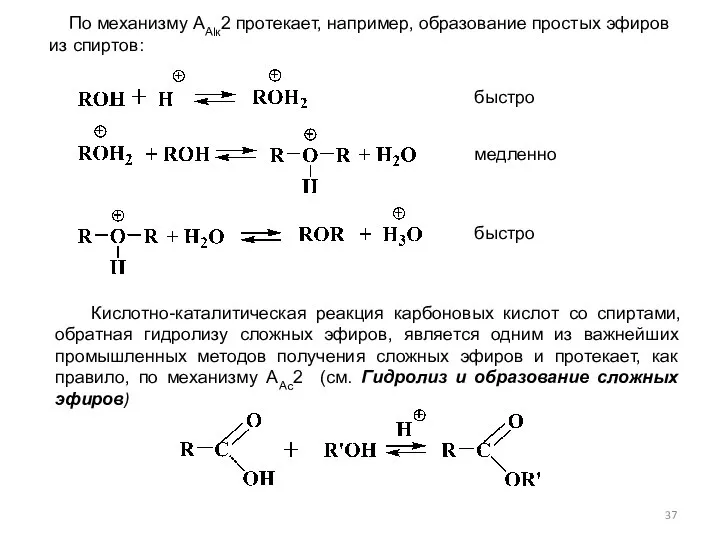
По механизму ААlк2 протекает, например, образование простых эфиров из спиртов:
По механизму ААlк2 протекает, например, образование простых эфиров из спиртов:
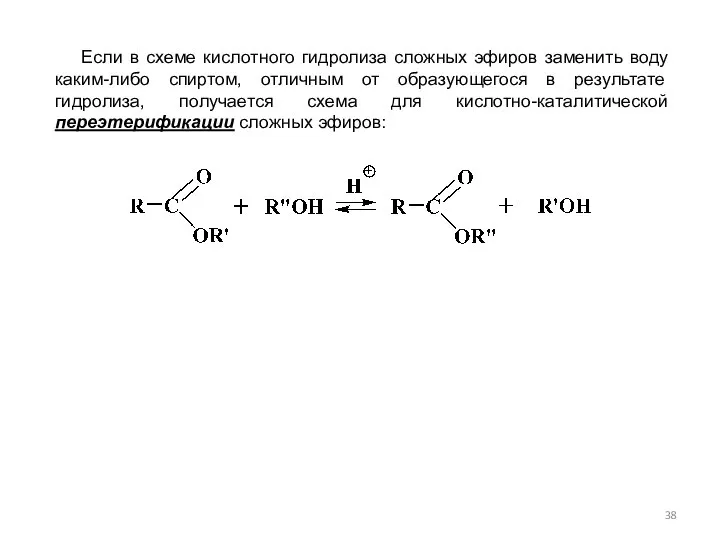
Если в схеме кислотного гидролиза сложных эфиров заменить воду каким-либо
Если в схеме кислотного гидролиза сложных эфиров заменить воду каким-либо
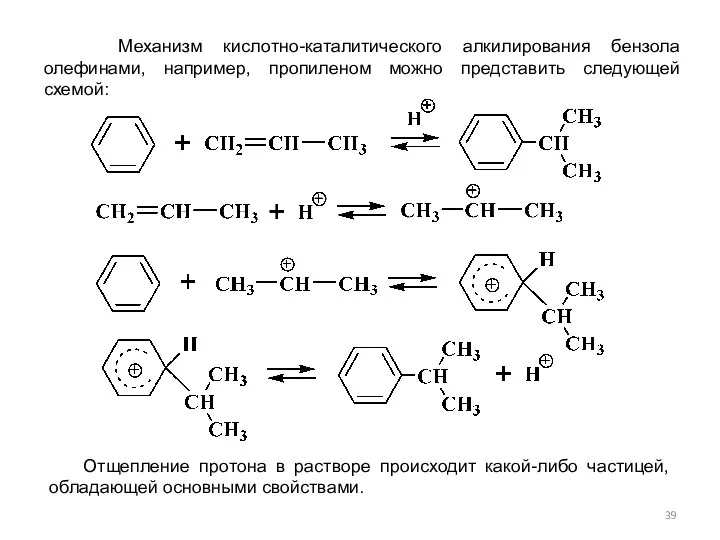
Механизм кислотно-каталитического алкилирования бензола олефинами, например, пропиленом можно представить следующей
Механизм кислотно-каталитического алкилирования бензола олефинами, например, пропиленом можно представить следующей

Электрофильный катализ или катализ апротонными кислотами
Заключается в активации реагента апротонной
Электрофильный катализ или катализ апротонными кислотами
Заключается в активации реагента апротонной

ОСНОВНОЙ КАТАЛИЗ
ОК - ускорение реакций в щелочной среде или в
ОСНОВНОЙ КАТАЛИЗ
ОК - ускорение реакций в щелочной среде или в
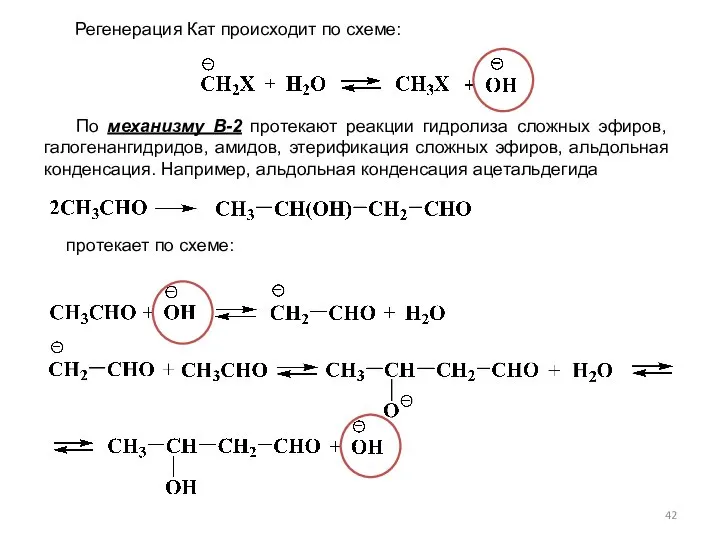
По механизму В-2 протекают реакции гидролиза сложных эфиров, галогенангидридов, амидов,
По механизму В-2 протекают реакции гидролиза сложных эфиров, галогенангидридов, амидов,

Нуклеофильный катализ – катализ основаниями, не связанный с кислотно-основным равновесием.
Нуклеофильный катализ – катализ основаниями, не связанный с кислотно-основным равновесием.

В сильно полярных растворителях устанавливается кислотно-основное равновесие:
НА + Н2О
В сильно полярных растворителях устанавливается кислотно-основное равновесие:
НА + Н2О

Различают специфический и общий кислотно-основной катализ (КОК).
Наблюдается, когда скорость
Различают специфический и общий кислотно-основной катализ (КОК).
Наблюдается, когда скорость
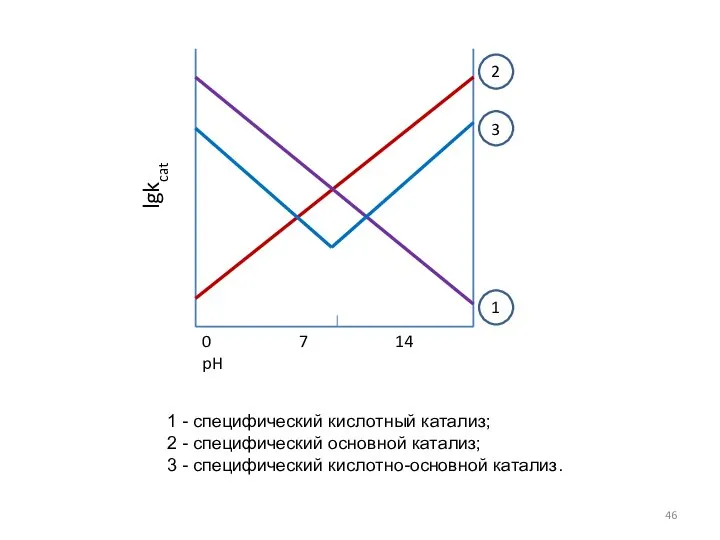
0 7 14 pH
lgkcat
1
2
3
1 - специфический кислотный катализ;
2 -
0 7 14 pH
lgkcat
1
2
3
1 - специфический кислотный катализ;
2 -

Осуществляется всеми кислотами и основаниями Бренстеда. Для определения этого катализа
Осуществляется всеми кислотами и основаниями Бренстеда. Для определения этого катализа

Недостатки гомогенного катализа:
необходимость отделения Кат от реакционной массы:
образование токсичных
Недостатки гомогенного катализа:
необходимость отделения Кат от реакционной массы:
образование токсичных



Разделы для самостоятельной проработки, 2013
Гомогенный металлокомплексный катализ.
Лит.
1. Лебедев Н.Н.,
Разделы для самостоятельной проработки, 2013
Гомогенный металлокомплексный катализ.
Лит.
1. Лебедев Н.Н.,
 Органічні розчинники. 11 клас
Органічні розчинники. 11 клас Электролитическая диссоциация воды
Электролитическая диссоциация воды Тема: «Бишофит – новый старый антигололедный реагент» Авторы: Гончаревич Анастасия Клокова Татьян
Тема: «Бишофит – новый старый антигололедный реагент» Авторы: Гончаревич Анастасия Клокова Татьян Алканы. Получение и применение алканов
Алканы. Получение и применение алканов Аминокислоты. Пептиды. Белки
Аминокислоты. Пептиды. Белки Значення хімічних процесів у природі
Значення хімічних процесів у природі H2O, Hg2+ CH3MgCI
H2O, Hg2+ CH3MgCI Карбонаты и фосфаты
Карбонаты и фосфаты Коллоидно-химический синтез квантовых точек на основе сульфидов свинца и кадмия
Коллоидно-химический синтез квантовых точек на основе сульфидов свинца и кадмия Микрофлора и биохимические реакции подземных вод
Микрофлора и биохимические реакции подземных вод Подгруппа мышьяка
Подгруппа мышьяка Комплексные соединения
Комплексные соединения Гормоны. Виды и классификация гормонов
Гормоны. Виды и классификация гормонов Предельные одноосновные карбоновые кислоты
Предельные одноосновные карбоновые кислоты Тұндыру әдістері
Тұндыру әдістері Самородные элементы
Самородные элементы Фармацевтическая химия
Фармацевтическая химия Презентация по Химии "Классификация углеводородов" - скачать смотреть
Презентация по Химии "Классификация углеводородов" - скачать смотреть  Получение и применение неметаллов
Получение и применение неметаллов Енергетичні напої користь чи шкода?
Енергетичні напої користь чи шкода?  Определение содержания растворенного кислорода в природной воде
Определение содержания растворенного кислорода в природной воде Геохимия природных процессов
Геохимия природных процессов Определение расхода воздуха на горение, количество и температуру продуктов
Определение расхода воздуха на горение, количество и температуру продуктов Влияние нанокомпозитов платина/Нафион на электрокаталитическую активность реакции восстановления водорода и окисления кислорода
Влияние нанокомпозитов платина/Нафион на электрокаталитическую активность реакции восстановления водорода и окисления кислорода Экспресс-методы решения задач по химии
Экспресс-методы решения задач по химии Неметаллы. Положение неметаллов в ПСХЭ Д.И. Менделеева. Галогены
Неметаллы. Положение неметаллов в ПСХЭ Д.И. Менделеева. Галогены Валентность. Составление формул по валентности
Валентность. Составление формул по валентности Дисперсные системы Эмульсии. Пены. Порошки
Дисперсные системы Эмульсии. Пены. Порошки