- Лекции по коллоидной химии

Содержание
- 2. Пособия: Фридрихсберг Д.А. Курс коллоидной химии. Л.: «Химия». 1984. 368 с. Щукин Е.Д., Перцов А.В., Амелина
- 3. Введение Основной объект изучения – дисперсные системы Дисперсная система - гетерогенная система, состоящая, как минимум, из
- 5. Дисперсная фаза и дисперсионная среда может иметь любое агрегатное состояние (Т, Ж, Г). В зависимости от
- 6. Наличие сильно развитой поверхности соприкосновения фаз разной физической или химической природы. Поверхность способна выполнять такие функции,
- 7. Общей геометрической особенностью всех дисперсных систем являются большая величина удельной поверхности контакта фаз (межфазной границы). Если
- 8. Величина Aо имеет так же смысл дисперсности измельченного вещества. Удельная поверхность характеризует дисперсную систему независимо от
- 10. Предельная адсорбция на единицу массы Xm (кмоль/кг) и на единицу площади Гm (кмоль/м2) связаны между собой
- 11. В коллоидных растворах удельная поверхность ( до 106 м2/кг ) и все ее функции достигают физически
- 12. 1.2 Адсорбция газов на поверхности твердых тел Теория мономолекулярной адсорбции Ленгмюра Это простейшая теория. В ней
- 13. В любой момент времени какая-то часть поверхности Θ занята адсорбированными молекулами, а оставшаяся часть (1 –
- 14. Каждая адсорбированная молекула с некоторой вероятностью kd может отрываться от поверхности адсорбента, т.е. десорбироваться. Скорость десорбции
- 15. Так что Θ = kaP/(kaP+ kd) (1.8) Объединив постоянные в константу адсорбционного равновесия k=ka/kd (1.9) получим:
- 16. Изотерма мономолекулярной адсорбции
- 17. Предельная адсорбция Γm может быть вычислена теоретически – по формуле (1.4) Гm=1/ s0 NA и экспериментально
- 18. Полимолекулярная адсорбция Толщина слоя адсорбированного вещества может быть больше размера молекул этого вещества. Адсорбционные силы это
- 19. Изотерма полимолекулярной адсорбции Поводом для создания теории послужил тот факт, что количество адсорбированного вещества может быть
- 20. Адсорбционный потенциал – работа переноса моля газа из бесконечности в адсорбционный объем. Так как газ конденсируется,
- 21. Характеристическая кривая
- 22. Теория БЭТ Адсорбция, как и в теории Ленгмюра, идет на активных центрах. Каждая адсорбированная молекула так
- 23. Поверхностное натяжение, поверхностный слой В математическом (геометрическом) смысле поверхность характеризуется величиной площади и кривизной. Это упрощенное
- 24. Поверхностный слой Поверхностный слой – часть гетерогенной системы, свойства которой отличаются от свойств обеих объемных фаз
- 25. Задача состоит в том, чтобы вычислить количество Ni i-того компонента во всей системе.
- 26. На схеме объемы фаз: v1=A(x0-0), v2=A(B-x0), v=A(B-0). В гипотетической системе концентрация Ci каждого компонента i неизменна
- 27. Что бы придать определенность величине адсорбции следует придерживаться определенной договоренности о положения разделяющей поверхности. Разделяющая поверхность,
- 28. 1.4 Поверхностное натяжение Поверхностное натяжение это работа образования единицы поверхности в обратимом изотермическом процессе. Или сила,
- 29. Молекулярный механизм натяжения Классическая трактовка
- 30. Молекулы поверхностного слоя втягиваются вглубь жидкой фазы всеми другим молекулам жидкой фазы. Поверхностное натяжение это работа
- 31. Приведенная выше трактовка природы поверхностного натяжения не согласуется с рядом фактов. Например, поверхностное натяжение воды можно
- 32. Механическая трактовка поверхностного натяжения как работы деформирования поверхностного слоя
- 33. Согласно определению, работа dW = ΣFidsi , где Fi – силы, действующие на грани параллелепипеда и
- 34. Работа деформирования поверхностного слоя это сумма (интеграл) работ деформирования тонких слоев: Рис. 1.7 Поверхностный слой толщиной
- 35. Согласно определению σ=W/dA , поэтому (1.25) Это формула Баккера. Натяжение существует потому, что соприкасающиеся фазы разделены
- 36. 3. Термодинамика поверхности Фундаментальное уравнение термодинамики выражает закон сохранения энергии: изменение dU энергии системы U равно
- 37. Уравнения термодинамики это обобщение уравнений механики и связанных с ними понятий: силы, координаты, энергии, работы. В
- 38. При наличии ряда компонентов суммируются вклады каждого в изменение химической составляющей энергии Σ μi dni .
- 39. Механическим аналогом уравнения (3.16) является условие равновесия рычага: Стрелы на схеме изображают действующие силы, обобщенные координаты
- 40. Для двухкомпонентного раствора: dσ = - Г1 dμ 1 - Г2 dμ 2 Это уравнение можно
- 41. Поверхностное натяжение растворов и адсорбция Двухкомпонентный раствор – смесь молекул двух разных веществ. Молекулы различаются полярностью
- 42. Адсорбция компонента это изменение его концентрации в поверхностном слое по сравнению с концентрацией в объемной фазе.
- 43. Адсорбция на границе раствор/воздух не поддается прямому измерению, поэтому на практике она вычисляется по Уравнению изотермы
- 44. Уменьшить натяжение раствора можно только путем введения в него вещества с меньшим натяжением, чем у растворителя,
- 45. ПАВ – вещества, способные изменять натяжение раствора при изменении их концентрации в растворе. В том числе:
- 46. Поверхностное натяжения раствора определяется составом поверхностного слоя. Если состав поверхностного слоя останется таким же как состав
- 47. По химическому строению ПАВ – дифильные вещества – состоят из полярной (гидрофильной) группы (-OH, -COOH, -NH3OH,
- 48. Способность вещества понижать натяжение характеризуется его поверхностной активностью: Графически это крутизна начального участка изотермы поверхностного натяжения
- 49. Поверхностная активность определяется соотношением сродства к воде полярной и неполярной частей дифильной молекулы. Количественно – эмпирической
- 50. Чем больше поверхностная активность, тем круче падение натяжения с ростом концентрации. Предельная величина натяжения одинакова для
- 51. Эмпирическая формула Шишковского: Аналитическое описание изотермы натяжения Δσ = σo- σ =Β ln (1+ kc) (4.1)
- 52. Изотерма адсорбции. Анализ уравнения Гиббса Рассмотренная выше на качественном уровне связь адсорбции и натяжения предполагает, что
- 53. Однако, в соответствии с уравнением Гиббса (3.1) – при большой концентрации раствора - в области постоянстве
- 54. Таким образом, одно и то же уравнение дает разные, взаимоисключающие зависимости (рис. 4.6 кривые 1 и
- 55. Причина несовпадения изотерм адсорбции (графиков 1 и 3), полученных разными способами обработки экспериментальной изотермы натяжения –
- 56. Правило Траубе для адсорбций Константа k изотермы Ленгмюра совпадает с одноименной константой уравнения Шишковского и, следовательно,
- 57. Полуколлоиды Полуколлоиды – вещества способные в зависимости от условий образовывать истинные или коллоидные растворы. К их
- 58. При ККМ мицеллы имеют сферическую форму. С увеличением концентрации ПАВ мицеллы становятся цилиндрическими и далее –
- 59. Применение мицеллярных растворов Солюбилизация – растворение нерастворимого. Мицеллярные растворы способны растворять вещества, которые в чистом растворителе
- 60. Строение адсорбционных слоев Опыты Ленгмюра Рис. 4.15 Нерастворимое в воде ПАВ, типа стеариновой кислоты, наносится на
- 61. В опытах при фиксированном и известном N изменятся площадь A, а весами измеряется сила F, действующая
- 62. Большой величине площади соответствует малая величине адсорбции N/A и малая степень заполнения поверхности Θ=Γ / Γm.
- 63. При сжатии адсорбционного слоя может наступить его конденсация – он становится несжимаемы – сопротивление сжатию резко
- 64. Опыты Ленгмюра впервые позволили измерить размеры молекул ПАВ – посадочную площадку Ao и их длину, поскольку
- 65. Капиллярные явления Капиллярные явления обусловлены кривизной границы раздела двух фаз. Следствие этих явлений: никакие нанообъекты с
- 66. Капиллярное давление это разность давлений в двух фазах, разделенных искривленной границей. На примере капли жидкости можно
- 67. Это сила поверхностного натяжения, пропорциональная длине окружности основания сегмента и сила разности давлений в капле и
- 68. Обобщение понятие радиуса в формуле Лапласа на случай несферической капли При произвольной форме капли (поверхности) формула
- 69. В равновесии давление в разных местах внутри замкнутой поверхности (и сосуда) одинаково. Если имеется несферичность капли,
- 70. Твердым веществам анизотропия присуща в силу их кристаллического строения, поэтому различные грани кристалла имеют разное по
- 71. Сплавы, керамика и другие кристаллические материалы - это поликристаллические вещества. В них имеются границы между соседними
- 72. 2.2 Капиллярные явления в трехфазных системах Пример трехфазной системы - капля жидкости (фаза 2 ) на
- 73. Общая линия соприкосновения трех фаз называется периметром смачивания. На схеме (рис. 2.2) она представлена точкой пересечения
- 74. Равновесная форма капли характеризуется величиной угла Θ. Он называется краевым углом смачивания Если Θ В обратном
- 75. Вещества, которые смачиваются водой, называются гидрофильными. Это вещества с ионным типом связи атомов, т.е. полярные с
- 76. Влияние кривизны поверхности на свойства веществ Переход одной из фаз гетерогенной системы в дисперсное состояние сопровождается
- 77. При равновесии на туже величину меняется потенциал и в газовой фазе. Полагая, что газ идеальный имеем
- 78. Капиллярное поднятие Обозначения: σ=σ23 , rk – радиус капилляра, r – радиус кривизны мениска. Классика: ρgh=2σ/rk
- 79. Адгезия и когезия адгезия W12 - удельная (на единицу поверхности контакта фаз) работа по разъединению фаз
- 80. Из определения натяжения как работы образования межфазной поверхности непосредственно следует: W12 = σ1 + σ2 -
- 81. При полном смачивании (Θ = 0 , cosΘ = 1 ) адгезия достигает своей максимально возможной
- 82. Адсорбция ПАВ на поверхности твердых веществ Все упомянутые выше результаты и зависимости, касающиеся адсорбции, в общем
- 83. Из водных растворов адсорбция идет на гидрофобных адсорбентах (сажа, графит, парафин, фторопласт). Из углеводородных растворителей адсорбция
- 84. Хемосорбция Хемосорбция – это адсорбция за счет химического взаимодействия полярной группы с поверхностью адсорбента. При хемосорбции
- 85. Адсорбция в металлах и сплавах Металлы и их сплавы – типичные представители поликристаллических материалов. Границы между
- 86. Тепловые эффекты и теплоты адсорбции и смачивания Адсорбенты – твердые вещества с большой величиной удельной поверхности
- 87. Тепловые эффекты Q dQ/dΓ Γ, m Рис. 4.11 Интегральный Q и дифференциальный dQ/dΓ тепловой эффект
- 88. Влияние температуры на адсорбцию следует из уравнения изотермы Гиббса По Гиббсу адсорбция монотонно падает с увеличением
- 89. Двойной электрический слой (ДЭС)
- 90. Образование и строение ДЭС ДЭС – система пространственно разделенных эквивалентных зарядов противоположного знака на границе раздела
- 92. Правило избирательной адсорбции ионов (правило Фаянса) Избирательно адсорбируются и заряжают поверхность те ионы, которые способны образовывать
- 93. Иначе говоря, знак и величина заряда поверхности определяется в этих особых случаях величиной pH раствора, т.е.
- 94. Из-за растворимости твердой фазы или диссоциации воды в растворе всегда присутствуют оба ПО иона данного вещества
- 95. Теория диффузного ДЭС Теория описывает строение внешней части ДЭС. Состояние внутренней части характеризуется потенциалом поверхности Ψs,
- 96. Количественно действие электрического поля характеризуется потенциальной энергией катионов z+FΨ и анионов z-FΨ в электрическом поле. Потенциал
- 97. Во внешней части ДЭС объемная плотность заряда ρ отлична от нуля благодаря различию концентраций противоионов и
- 98. Распределение в пространстве заряда и потенциала подчиняется фундаментальному закону электростатики -закону Пуассона. Если потенциал зависит только
- 99. Другая форма левой части уравнения (5.7) d(dΨ/dx)/dx. Так что после умножения обех частей на dΨ оно
- 100. Ψ= Ψs exp(-æx) (5.9) Уравнение показывает, что ДЭС устроен диффузно - не имеет четкой внешней границы
- 101. Интегрирование уравнения Пуассона дает заряд qv внешней части ДЭС (5.10) По условию электронейтральности ДЭС его поверхностный
- 102. Развитие теории ДЭС Электрическая емкость ДЭС доступна прямому измерению – это емкость электролитических конденсаторов. Установлено, что
- 103. Строение ДЭС с учетом размера ионов Рис. 5.3
- 104. Соответствующее распределение потенциала Рис. 5,4
- 105. Фактически показанное на рис. (5.3 и 5.4) распределение ионов и потенциала реализуется только при наличии дополнительных
- 106. Влияние электролитов на строение ДЭС В рамках изложенных представлений ДЭС исчерпывающе характеризуется толщиной δ=1/æ , потенциалом
- 107. Распределение потенциала Ψ Ψs ζ1 ζ2 ζ3=0 x c1 Рис. 5.6 Сжатие ДЭС электролитом: уменьшение толщины
- 108. Влияние электролитов на ζ-потенциал Сжатие ДЭС электролитом вызывает уменьшение ζ-потенциала, вплоть до нуля и, далее, до
- 109. Действие ПО электролитов на ДЭС Представленный выше вариант теории, как и более полный – при произвольной
- 110. Электрокапиллярность Электрокапиллярные эффекты – зависимость поверхностного натяжения межфазной границы от разности электрических потенциалов фаз. Они исследованы
- 111. Электрокапиллярная кривая – график зависимости натяжения границы ртуть/вода от величины внешнего отрицательного потенциала, поданного на ртуть
- 112. Уравнение электрокапиллярной кривой – уравнение Липпмана: qs= - dσ/dU (5.15) На левой ветви кривой отрицательный потенциал
- 113. В формуле (5.16) присутствуют заряд и потенциал. Они могут быть связаны (см. (5.12)) прямой пропорциональной зависимостью
- 114. 2.3 Пленки Пленка – это часть фазы, ограниченная с двух сторон границами с другой фазой (симметричная
- 115. Пленки делятся на толстые и тонкие. Толстая пленка имеет толщину h большую, чем сумма толщин δ
- 116. Пленка, которая стремится к самопроизвольному увеличению толщины создает положительное расклинивающее давление (П> 0) на ограничивающие ее
- 117. Сближение тел, ограничивающих толщину пленки, требует совершения работы w против силы расклинивающего давления. При сближении от
- 118. Устойчивость дисперсных систем Коллоидные растворы (и другие дисперсные системы) термодинамически неустойчивы – имеют тенденцию к самопроизвольному
- 119. Если таких мер не предпринимать, то во взаимодействии частиц будет преобладать притяжение и они будут слипаться.
- 120. В вакууме, и в газовой среде молекулярные силы всегда ведут к взаимному притяжению двух частиц, поскольку
- 121. Иначе говоря, жидкая дисперсионная среда, обладающая хорошим сродством к поверхности частиц (адгезией), втягивается в зазор между
- 122. Отталкивание может иметь разную природу. Природа сил отталкивания (факторы агрегативной устойчивости): электростатическая – отталкивание двойных электрических
- 123. Электростатическое отталкивание ДЭС (Электростатическая составляющая расклинивающего давления) Между двумя телами, разделенными пленкой жидкости толщиной h, действует
- 124. ДЭС тонкой пленки Потенциал в зазоре – сплошная кривая на рис. 6.1. Это результат наложения полей
- 125. Распределение ионов и потенциала в зазоре подчиняется тем же законам, что и возле свободной поверхности и
- 126. После звлечения корня квадратного имеем: dΨ/dx= æ (Ψ2 - Ψh2)1/2. Интегрируя это уравнения от Ψh до
- 127. В формуле (6.2) потенциал Ψh нужно заменить не зависящим от h потенциалом поверхности Ψs. Так как
- 128. Производная функции (6.3): dΨ/dx = æΨssh(æx) / ch(æh/2). При x= - h/2 (dΨ/dx)s = - æΨssh(æh/2)
- 129. Молекулярное притяжение Расчет энергии молекулярного притяжения двух плоских тел, разделенных узким зазором h, основан на принципе
- 130. Принцип аддитивности Гамакера Тело 2 Рис. 6.2 К выводу формулы энергии молекулярного притяжения тел 1 и
- 131. Выберем (рис. 6.2) две произвольные молекулы 1 в теле 1 и 2 в теле 2. Расстояние
- 132. Все тело 2 представимо как совокупность сферических слоев (половины качана капусты). Взаимодействие молекулы 1 с каждым
- 133. Тело 1 представимо как совокупность тонких плоских слоев толщиной dx. Объем одного такого слоя с площадью
- 134. Та же формула в компактной записи Uм= - AH / h2 (6.11) AH= (π/12)(NA/Vm)2A1 – константа
- 135. Теория взаимодействия фаз Лифшица По Лифшицу тела взаимодействуют между собой своими электромагнитными полями. Неотъемлемой принадлежностью любого
- 136. Динамические поля тел, в отличие от статического поля ДЭС, создают силы их взаимного притяжения, поскольку поля
- 137. Выполнение соответствующих расчетов требует задания набора частот, амплитуд и фаз электромагнитных колебаний каждого из взаимодействующих тел,
- 138. Учет эффекта электромагнитного запаздывания ведет к тому, что теория Лифшица дает разные выражения для энергии взаимодействия
- 139. Теория ДЛФО
- 140. Переход Дерягина Для объяснения свойств коллоидных растворов необходимо анализировать взаимодействие сферических частиц, тогда как имеются только
- 142. На поверхности сфер радиуса a выделим симметричные пояски шириной dh. Положим, что они взаимодействуют между собой
- 143. Подставляя в (7.1) выражение (6.6) для энергии отталкивания плоских заряженных тел, получим формулу для расчета энергии
- 144. При произвольной величине потенциала Ψs и соблюдении всех других ограничений (æh >1, a >h) действует та
- 145. При неограниченном увеличении потенциала функция γ (7.5) стремится к единице. Таким образом, максимально возможная величина константы
- 146. Потенциальные кривые и устойчивость В теории ДЛФО состояние и поведение дисперсной системы определяется суммарной величиной U
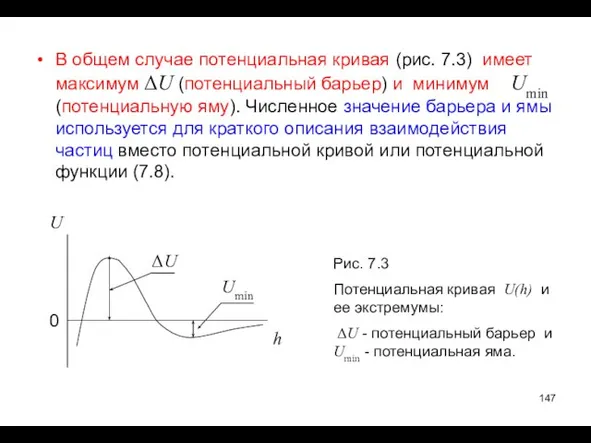
- 147. В общем случае потенциальная кривая (рис. 7.3) имеет максимум ΔU (потенциальный барьер) и минимум Umin (потенциальную
- 148. Энергия взаимодействия частиц сложным образом зависит от расстояния между ними: на расстояниях близких к нулю всегда
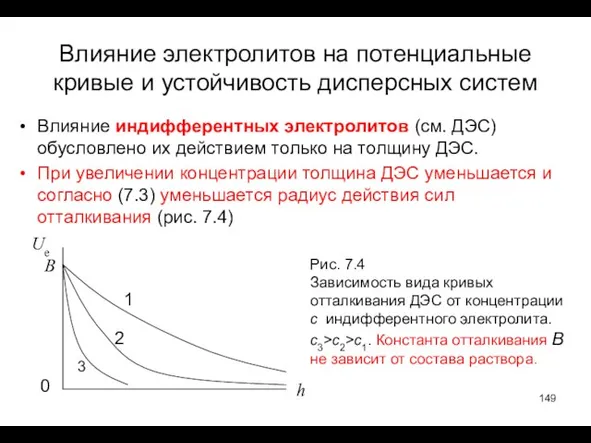
- 149. Влияние электролитов на потенциальные кривые и устойчивость дисперсных систем Влияние индифферентных электролитов (см. ДЭС) обусловлено их
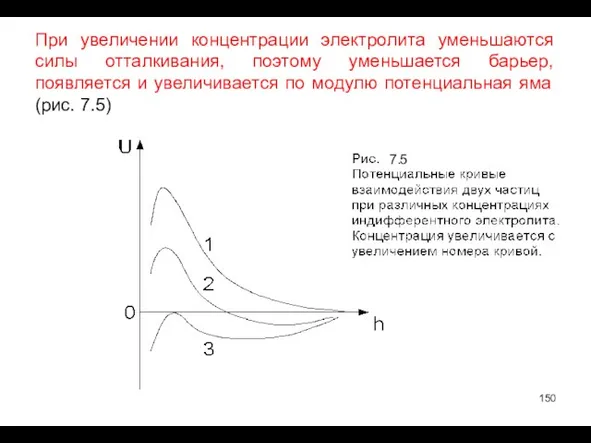
- 150. При увеличении концентрации электролита уменьшаются силы отталкивания, поэтому уменьшается барьер, появляется и увеличивается по модулю потенциальная
- 151. В коллоидных системах, как и в молекулярных, изменение состояния системы осуществляется с помощью универсального механизма -
- 152. 1. ΔU >> kBT, [Umin] При таком соотношении экстремумов функции взаимодействия и средней энергии столкновений происходят
- 153. 2. ΔU ≥ kBT, [Umin] При сопоставимости величин энергии столкновений и барьера возможно преодоление барьера с
- 154. 3. ΔU >> kBT, [Umin] ≥ kBT. - большой барьер и достаточно большая потенциальная яма (рис.7.5-2).
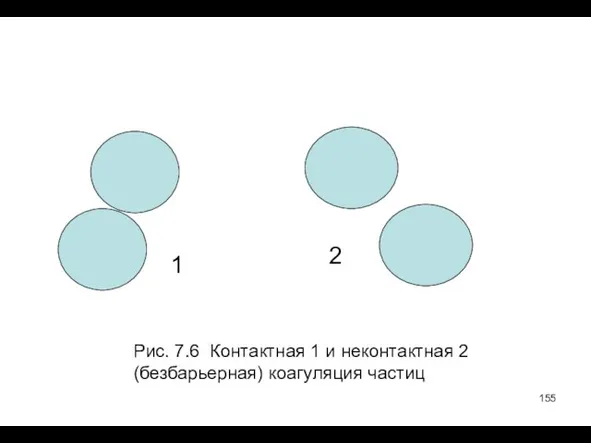
- 155. 1 2 Рис. 7.6 Контактная 1 и неконтактная 2 (безбарьерная) коагуляция частиц
- 156. 4. ΔU = 0 - потенциальный барьер равен нулю. При таком типе взаимодействия частиц полностью отсутствует
- 157. Влияние неиндифферентных (потенциалопределяющих) электролитов (см. ДЭС) связано с их действием на потенциал поверхности и, следовательно, на
- 158. Рис. 7.7 Влияние ПО электролита на энергию отталкивания ДЭС. С увеличением концентрации одноименного ПО иона константа
- 159. Изменение сил отталкивания под действием ПО электролита в общем вызывает такие же изменения суммарной потенциальной кривой,
- 160. Критическая концентрация электролита и правила электролитной коагуляции Условие быстрой необратимой коагуляции ΔU=0 в развернутом виде: B
- 161. Из структуры формул первой и второй строки следует, что эти обе формулы (7.9) совместимы если æ
- 162. Частные виды формулы (7,12) При большом потенциале поверхности, согласно (7.6) с точностью до постоянных множителей B*
- 163. Закон (7.13) действует только при концентрационной коагуляции индифферентными электролитами. Закон (7.14) действует при нейтрализационной коагуляции растворов
- 164. Эффект смещения плоскости локализации заряда относительно межфазной границы Не существует физически обоснованных причин считать, что плоскость
- 165. Правила электролитной коагуляции, сопоставление с теорией ДЛФО Правила коагуляции это результат обобщение разнообразных по объектам и
- 166. 3 – правило альтернативное правилу 2. Коагуляция наступает при уменьшении абсолютной величины электрокинетического потенциала частиц до
- 167. Первое правило объясняется тем, что любой электролит при возрастании концентрации увеличивает параметра Дебая æ, доводя его
- 168. Получение дисперсных систем Получение дисперсной системы сводится к решению двух задач: Получению частиц малого размера и
- 169. При химической конденсации (образование частиц AgI в растворе, дыма при горении топлива) пересыщение создается проведением химической
- 170. Критический зародыш – минимальный по размеру зародыш новой фазы, который при случайном возникновении в пересыщенной в
- 171. Химическая конденсация Правила образования коллоидного раствора непосредственно при проведении химической реакции: 1. Реакция ведется при избытке
- 172. Метод дробления. Эффект Ребиндера. Дроблением получают дисперсные системы с размером частиц 10-6 м и более. При
- 173. Пептизация Пептизация – получение коллоидных растворов путем разрыхления осадков и перевода взвеси в агрегативно устойчивое состояние.
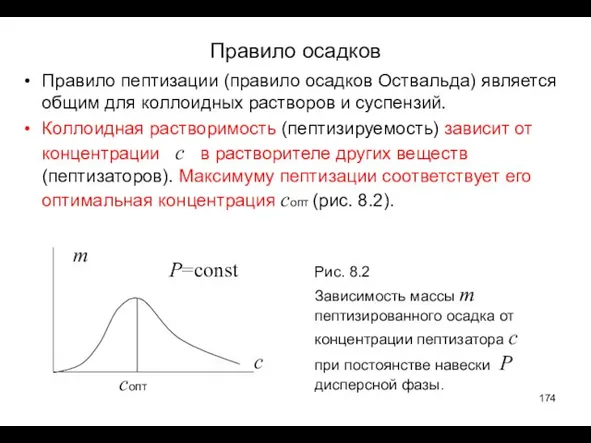
- 174. Правило осадков Правило пептизации (правило осадков Оствальда) является общим для коллоидных растворов и суспензий. Коллоидная растворимость
- 175. С позиций теории ДЛФО пептизатор это ПО электролит. Увеличение его концентрации увеличивает потенциал поверхности, константу отталкивания
- 176. Молекулярно-кинетические свойства дисперсных систем
- 177. Седиментация и диффузия Молекулярно кинетические свойства и явления это явления обусловленные движением частиц. Существую два типа
- 178. Уникальность коллоидов в том, что только в них интенсивность обоих типов движения сопоставима по величине: в
- 179. Формула Эйнштейна для коэффициента диффузии: D=kT/6πηa (9.2) 6πηa - коэффициент Стокса – сила сопротивления движению частицы
- 180. Масса частицы m=vρ, где v =4πa3/3 – объем частицы и ρ – плотность дисперсной фазы. С
- 181. Седиментационно-диффузионное равновесие Одновременное участие частиц в оседании и диффузии приводит к установлению во взвеси седиментационно-диффузионного равновесия.
- 182. Уравнение седиментационно-диффузионного равновесия n=n0 exp( -mgh/ kBT) (9.6) По сути это закон распределения Больцмана по потенциальным
- 183. Нормирование распределения Больцмана Обычно известна средняя по высоте (рецептурная) концентрация дисперсной фазы n=N /V, где N
- 184. Формула (9.7) выражает условие нормирования распределения Больцмана - нахождения неизвестной константы распределения n0 После интегрирования находим
- 185. Рис. 9.1
- 186. Высота осадка находится из условия сохранения вещества при разделении системы на осадок и взвесь: N =
- 187. Электрокинетические явления Это явления связанные со взаимным смещением фаз дисперсной системы. Прямые электрокинетические явления: 1 Электрофорез
- 188. Электрофорез и потенциал оседания возникают в относительно разбавленных дисперсных системах, где частицы имеют возможность перемещаться относительно
- 189. Рис. 9.2 Схема 1 – электрофорез Схема 2 - электроосмос
- 190. Механизм электрофореза Не тривиальность явления электрофореза обусловлена тем, что в растворе электролита заряженные частицы всегда окружены
- 191. Механизм фореза Рис. 9.3 Деформация и релаксация ДЭС при электрофорезе. E – силовые линии поля диполя.
- 192. Ионы раствора притягиваются к противоположным по знаку полюсам диполя, достраивая недостающую часть ионной атмосферы на одном
- 193. Электроосмос Рис. 9.4 Схема электроосмотического течения в капилляре
- 194. Пористое тело это совокупность параллельных потоку капилляров. Схема рис. 9.4 представляет один из них. На внутренних
- 195. Вывод формулы для скорости фореза и осмоса На примере электроосмоса. На схеме рис. 9.5 для простоты
- 196. Силы, действующие внутри ДЭС на слой жидкости толщиной dx и площадью большой грани равной единице: Электрическая
- 197. Результирующая сила трения fv равна разнице сил трения среды о внешнюю и внутреннюю грани слоя, т.е.
- 198. Второе интегрирование - (после умножения на dx) от x=d где Ψ=ζ и u=0 до x=∞ ,
- 199. Объемная скорость электроосмоса v=us, где s - площадь поперечного сечения всех капилляров пористой перегородки. При электроосмосе
- 200. Обратные явления Потенциал оседания + -
- 201. Потенциал протекания При положительном заряде частиц + -
- 202. Кинетика коагуляции Теория ДЛФО формулирует условие быстрой необратимой коагуляции, а кинетику этого процесса описывает теория Смолуховского:
- 203. При R ≤ Rk , т.е. при столкновении, вступает в действие большая сила притяжения и частицы
- 204. Составная частица из нескольких слипшихся первичных частиц называется флокулой Формула (9.17) дает общую счетную концентрацию всех
- 205. Удобной характеристикой текущего состояния коагулирующей системы является среднее число частиц в одной флокуле Среднее число частиц
- 206. Теория Смолуховского дает и более детальное описание процесса коагуляции, а именно, зависимость от времени концентрации флокул
- 207. Оптические свойства дисперсных систем Прохождение света через любую оптическую среду сопровождается его ослаблением (или усилением в
- 208. Соответственно, коэффициент ослабления K складывается из коэффициента поглощения Kп и коэффициента рассеяния Kр : K=Kп+Kр Поглощение,
- 209. Электрическое поле световой волны индуцирует в частицах дипольный момент, который осциллирует с частотой падающей на частицу
- 210. 10.1
- 211. Диаграммы рассеяния естественного света получается суперпозицией (суммой) множества диаграмм, получаемых вращением диаграммы типа 10.1 вокруг оси
- 212. Рис. 10.3 Конус Тиндаля 1 2 3 4
- 213. Структура и структурирование дисперсных систем Структура – характеристика взаимного расположения частиц. Структурирование – фиксация взаимного расположения
- 214. Рис. 11.1 Не структурированная 1 и коагуляционно структурированная 2 система 1 2 t τ
- 215. Наиболее распространено коагуляционное структурирование, обычно при обратимой безбарьерной коагуляции (ΔU>>kT, Umin>kT). Результатом является образование рыхлой сетки
- 216. Фрактальная размерность Рис. 11.2. Флокула r L ν=(L / r)ф ν=1+t/t* t*=3η/8kTnн
- 217. Фрактальная размерность (рис. 11.2) – показатель степени ф, определяющий зависимость размера флокулы L от числа частиц
- 218. Минимально возможное число связанных частиц во флокуле реализуется, если они расположены цепочкой по диаметру флокулы (рис.
- 219. Коагуляционное структурирование В процессе коагуляции среднее число частиц в одной флокуле растет согласно уравнению кинетики коагуляции
- 220. Крупные флокулы более рыхлые, чем мелкие и потому менее прочные. В потоке они обратимо разрушаются на
- 221. Структурирование в поле обусловлено дипольным взаимодействием частиц UII и UT. Оно обратимо: при выключении поля структура
- 222. Структурирование устойчивых систем за счет ограниченности объема (ϕ≥0.52) В условиях отсутствия свободного пространства частицы за счет
- 223. Прочность (энергия ΔU) фиксации частиц за счет отталкивания (на примере потенциальных кривых взаимодействия трех частиц) Рис.
- 224. Реология Наука о деформационных свойствах материалов Напряжение сдвига τ =F / A, деформация сдвига γ =δL
- 225. Ньютоновская жидкость η = const (12.2-1) Неньютоновская η = f(τ) (12.2-2) Рис. 12.2. Реологические кривые ньютоновской
- 226. Пластичность ПЛАСТИЧНОСТЬ – способность к неограниченным по величине деформациям без разрушения материала. В этом сходство с
- 227. Принадлежность к тому или иному типу определяется структурой системы Ньютоновские – неструктурированные Вязкость ньютоновской жидкости описывается
- 228. Реология тиксотропных систем Применение формул Эйнштейна-Бринкмена к флокулированным системам η=η0 / (1- ϕe)α (12.6) ϕe=1 при
- 229. Рис. 12.3. Реологические кривые течения γ’ и вязкости η согласно (12.6): γ’ τ τs τm 0
- 230. Рис. 12.4. Полные реологические кривые (ПРК) I II III γ’ η 0 τd τm τ Кривые
- 231. Реология ПКС Рис. 12.5. Течение ПКС (слайд 222) без увеличя объема системы (дилатации) возможно при наличии
- 232. Реологическое уравнение течения ПКС при малых напряжениях (график на рис. 12.6): γ’=(c/n) τ/ηe(1+trτ/ηe) -1 n –
- 233. Реодинамика – гидродинамика неньютоновых жидкостей Течение в трубе ньютоновской жидкости. u - линейная (м/с), v -
- 234. Течение в трубе пластичной жидкости τ = τs + η*γ’ Рис. 18.8 Расход v, как функция
- 235. Вязкоупругость. Вязкоупругое твердое тело Рис. 12.9 Аддитивны напряжения τ=Gγ+ηγ’ , Релаксация деформации при τ=0: Gγ=-ηdγ/dt, dγ/γ=-(G/η)dt,
- 236. Вязкоупругая жидкость Рис. 12.10 Аддитивны деформации : γ = γG + γN Релаксация напряжений при γ
- 237. Дисперсные системы
- 238. Суспензии Проблема – обеспечение однородности при минимальной концентрации Диагностика: V t 0 V∞ V∞ 1 2
- 239. Загустители – структурообразователи Полидисперсность. Umin~(a1a2)1/2 При максимальной концентрации – ПКС Ограниченное время текучести ПКС Рис. 13.2
- 240. Эмульсии Грубодисперсные системы типа Ж1/Ж2 (вода и масло) 1. Разбавленные, ϕ 2. Концентрированные, ϕ=1-75% 3. Высококонцентрированные
- 241. 1 могут существовать без стабилизатора, во 2 и 3 обязательно наличие стабилизатора (эмульгатора) Прямые - М/В
- 242. М М В В В М RCOONa (RCOO)2Ca
- 243. М М М В В В В Жидкая фаза высококонцентрированных эмульсий это тонкие пленки ПАВ:
- 244. Получение эмульсий - диспергированием смеси жидкостей в присутствии ПАВ Обращение фаз М/В↔В/М: RCOONa +CaCl2→(RCOO)2Ca + NaCl
- 245. Системы типа Т1/Т2 Металлы и сплавы Керамика Наполненные полимеры (пластмассы, резина) Минеральные ископаемые Искусственные камни (бетон)
- 246. Системы типа Г/Ж и Г/Т Аналог прямых эмульсий, газ вместо масла Высококонцентрированные системы типа Г/Ж и
- 247. Аэрозоли системы типа Т/Г и Ж/Г Т/Г – дым, пыль, Т/Ж – туман Получаются конденсацией (дым,
- 248. Размерные эффекты, нанотехнологии Зависимость физических свойств от размера частиц и толщины пленок Формула Томсона, расклинивающее давление
- 249. Полимеры
- 250. Строение и тепловое движение Полиэтилен и производные Метан a , этан b , пропан c. Угол
- 251. Специфичный тип теплового движения – вращение звеньев цепи около оси С-С Это придает полимерной цепи гибкость
- 252. Возможность вращения звеньев определяется потенциальным барьером вращения ΔUr: ΔUr
- 253. Наиболее вероятная форма молекулы Микро и макросостояния L - характеристика макросостояния Sk=k lnw, w – термодинамическая
- 254. Свободная энергия F = U - TSk Потенциальный барьер, вращательная подвижность звеньев и Sk определяются полярностью
- 255. Эластичность –способность к большим обратимым деформациям Механизм эластичности: Механизм пластичности: Пластификатор и вулканизатор
- 256. Кристаллизация, роль боковых цепочек
- 257. Термомеханическая кривая Зависимость деформации γ от температуры T при постоянстве нагрузки 1 – стекловидность 2 –
- 258. Растворы полимеров Сходство и различие с коллоидами Условие растворения ΔG Для полярных ΔSk=0, ΔH Для неполярных
- 259. Степень набухания ΔV/V0 a – ограниченное, b - с частичным растворением, c - неограниченное ΔV/V0 0
- 260. Вязкость растворов В гидродинамическом отношении разбухшие клубки - сплошные частиц, применим вариант формулы Эйнштейна η=η0(1+αϕ+βϕ2) или
- 261. Слагаемое βϕ2 в формуле Эйнштейна – учет гидродинамического взаимодействия клубков. Это полуразбавленные растворы. В концентрированных цепи
- 262. Полиэлектролиты - полимеры, содержащие звенья, способные диссоциировать в растворе. Поликислоты, полиоснования и амфотерные полиэлектролиты. Желатин –
- 263. Изоэлектрическая точка (ТНЗ) pH q 0 + -
- 264. Вдали от ТНЗ отталкивание одноименно заряженных звеньев ведет к разбуханию клубка. В ТНЗ цепь имеет эквивалентное
- 265. Соответственно влиянию рН на заряд и размер клубков изменяются свойства растворов: вязкость, степень набухания: η, ΔV/
- 267. Скачать презентацию

Пособия:
Фридрихсберг Д.А. Курс коллоидной химии.
Л.: «Химия». 1984. 368
Пособия:
Фридрихсберг Д.А. Курс коллоидной химии.
Л.: «Химия». 1984. 368

Введение
Основной объект изучения – дисперсные системы
Дисперсная система - гетерогенная система,
Введение
Основной объект изучения – дисперсные системы
Дисперсная система - гетерогенная система,
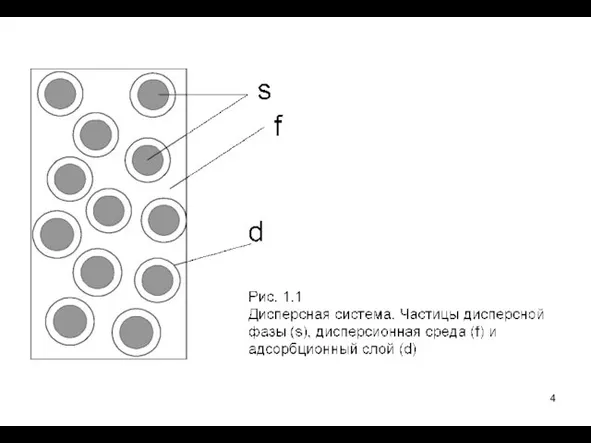

Дисперсная фаза и дисперсионная среда может иметь любое агрегатное состояние (Т,
Дисперсная фаза и дисперсионная среда может иметь любое агрегатное состояние (Т,

Наличие сильно развитой поверхности соприкосновения фаз разной физической или химической природы.
Наличие сильно развитой поверхности соприкосновения фаз разной физической или химической природы.

Общей геометрической особенностью всех дисперсных систем являются большая величина удельной поверхности
Общей геометрической особенностью всех дисперсных систем являются большая величина удельной поверхности

Величина Aо имеет так же смысл дисперсности измельченного вещества.
Удельная поверхность характеризует
Величина Aо имеет так же смысл дисперсности измельченного вещества.
Удельная поверхность характеризует

Предельная адсорбция на единицу массы Xm (кмоль/кг) и на единицу площади
Предельная адсорбция на единицу массы Xm (кмоль/кг) и на единицу площади

В коллоидных растворах удельная поверхность ( до 106 м2/кг ) и
В коллоидных растворах удельная поверхность ( до 106 м2/кг ) и

1.2 Адсорбция газов на поверхности твердых тел
Теория мономолекулярной адсорбции Ленгмюра
Это простейшая
1.2 Адсорбция газов на поверхности твердых тел
Теория мономолекулярной адсорбции Ленгмюра
Это простейшая

В любой момент времени какая-то часть поверхности Θ занята адсорбированными молекулами,
В любой момент времени какая-то часть поверхности Θ занята адсорбированными молекулами,

Каждая адсорбированная молекула с некоторой вероятностью kd может отрываться от поверхности
Каждая адсорбированная молекула с некоторой вероятностью kd может отрываться от поверхности

Так что
Θ = kaP/(kaP+ kd) (1.8)
Объединив постоянные в константу адсорбционного равновесия
Так что
Θ = kaP/(kaP+ kd) (1.8)
Объединив постоянные в константу адсорбционного равновесия
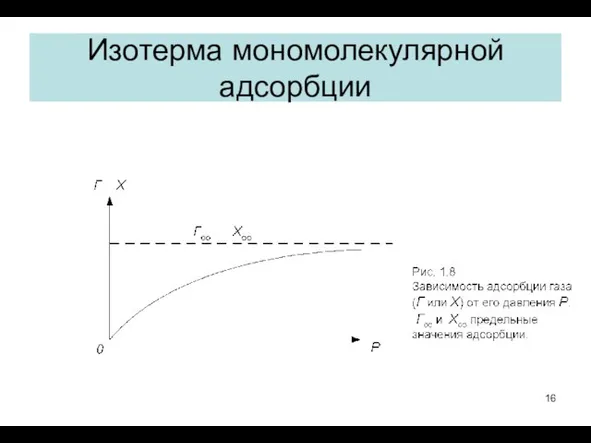
Изотерма мономолекулярной адсорбции
Изотерма мономолекулярной адсорбции

Предельная адсорбция Γm может быть вычислена теоретически – по формуле (1.4)
Предельная адсорбция Γm может быть вычислена теоретически – по формуле (1.4)

Полимолекулярная адсорбция
Толщина слоя адсорбированного вещества может быть больше размера молекул этого
Полимолекулярная адсорбция
Толщина слоя адсорбированного вещества может быть больше размера молекул этого
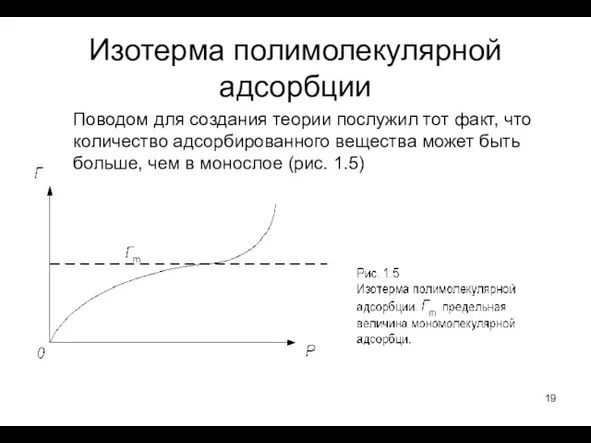
Изотерма полимолекулярной адсорбции
Поводом для создания теории послужил тот факт, что количество
Изотерма полимолекулярной адсорбции
Поводом для создания теории послужил тот факт, что количество

Адсорбционный потенциал – работа переноса моля газа из бесконечности в адсорбционный
Адсорбционный потенциал – работа переноса моля газа из бесконечности в адсорбционный
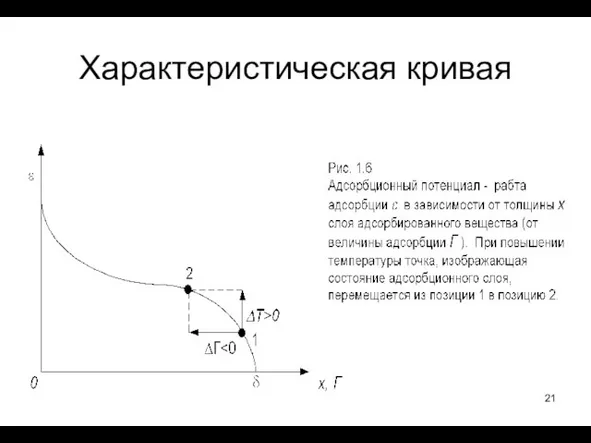
Характеристическая кривая
Характеристическая кривая

Теория БЭТ
Адсорбция, как и в теории Ленгмюра, идет на активных центрах.
Каждая
Теория БЭТ
Адсорбция, как и в теории Ленгмюра, идет на активных центрах.
Каждая

Поверхностное натяжение, поверхностный слой
В математическом (геометрическом) смысле поверхность характеризуется величиной площади
Поверхностное натяжение, поверхностный слой
В математическом (геометрическом) смысле поверхность характеризуется величиной площади
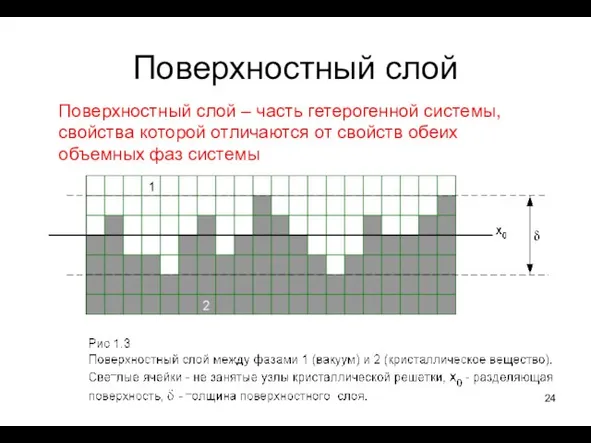
Поверхностный слой
Поверхностный слой – часть гетерогенной системы, свойства которой отличаются от
Поверхностный слой
Поверхностный слой – часть гетерогенной системы, свойства которой отличаются от
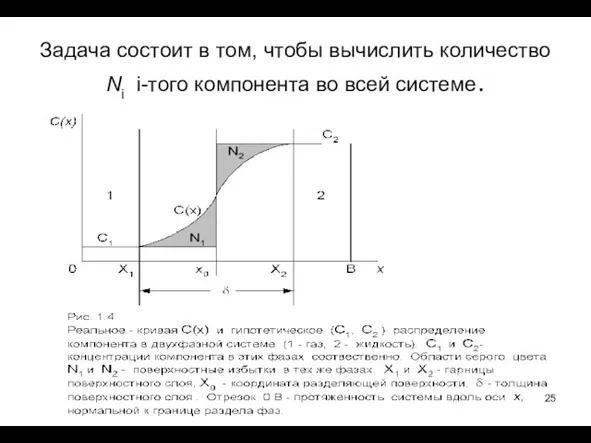
Задача состоит в том, чтобы вычислить количество Ni i-того компонента во
Задача состоит в том, чтобы вычислить количество Ni i-того компонента во

На схеме объемы фаз:
v1=A(x0-0), v2=A(B-x0), v=A(B-0).
В гипотетической системе концентрация Ci
На схеме объемы фаз:
v1=A(x0-0), v2=A(B-x0), v=A(B-0).
В гипотетической системе концентрация Ci

Что бы придать определенность величине адсорбции следует придерживаться определенной договоренности о
Что бы придать определенность величине адсорбции следует придерживаться определенной договоренности о

1.4 Поверхностное натяжение
Поверхностное натяжение это работа образования единицы поверхности в
1.4 Поверхностное натяжение
Поверхностное натяжение это работа образования единицы поверхности в
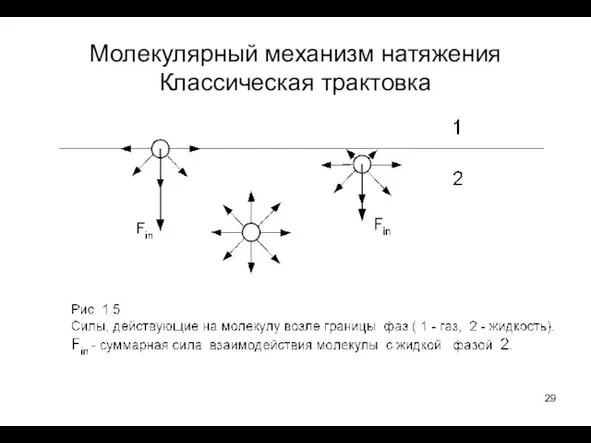
Молекулярный механизм натяжения
Классическая трактовка
Молекулярный механизм натяжения
Классическая трактовка

Молекулы поверхностного слоя втягиваются вглубь жидкой фазы всеми другим молекулам жидкой
Молекулы поверхностного слоя втягиваются вглубь жидкой фазы всеми другим молекулам жидкой

Приведенная выше трактовка природы поверхностного натяжения не согласуется с рядом фактов.
Приведенная выше трактовка природы поверхностного натяжения не согласуется с рядом фактов.
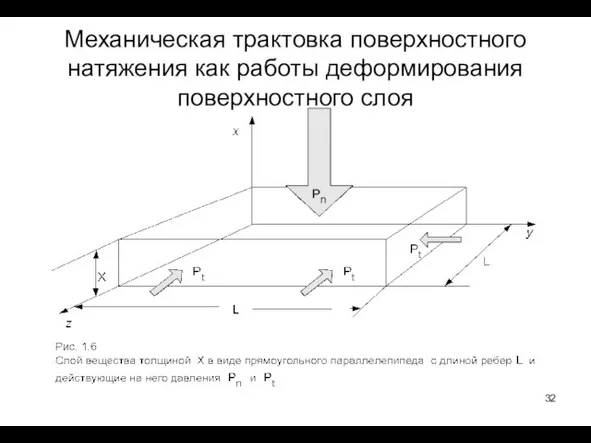
Механическая трактовка поверхностного натяжения как работы деформирования поверхностного слоя
Механическая трактовка поверхностного натяжения как работы деформирования поверхностного слоя

Согласно определению, работа dW = ΣFidsi , где Fi – силы,
Согласно определению, работа dW = ΣFidsi , где Fi – силы,

Работа деформирования поверхностного слоя это сумма (интеграл) работ деформирования тонких слоев:
Рис.
Работа деформирования поверхностного слоя это сумма (интеграл) работ деформирования тонких слоев:
Рис.

Согласно определению σ=W/dA , поэтому
(1.25)
Это формула Баккера. Натяжение существует потому, что
Согласно определению σ=W/dA , поэтому
(1.25)
Это формула Баккера. Натяжение существует потому, что

3. Термодинамика поверхности
Фундаментальное уравнение термодинамики выражает закон сохранения энергии:
изменение dU
3. Термодинамика поверхности
Фундаментальное уравнение термодинамики выражает закон сохранения энергии:
изменение dU

Уравнения термодинамики это обобщение уравнений механики и связанных с ними понятий:

При наличии ряда компонентов суммируются вклады каждого в изменение химической составляющей
При наличии ряда компонентов суммируются вклады каждого в изменение химической составляющей

Механическим аналогом уравнения (3.16) является условие равновесия рычага:
Стрелы на схеме изображают
Механическим аналогом уравнения (3.16) является условие равновесия рычага:
Стрелы на схеме изображают

Для двухкомпонентного раствора:
dσ = - Г1 dμ 1 - Г2 dμ
Для двухкомпонентного раствора:
dσ = - Г1 dμ 1 - Г2 dμ

Поверхностное натяжение растворов и адсорбция
Двухкомпонентный раствор – смесь молекул двух разных
Поверхностное натяжение растворов и адсорбция
Двухкомпонентный раствор – смесь молекул двух разных

Адсорбция компонента это изменение его концентрации в поверхностном слое по сравнению
Адсорбция компонента это изменение его концентрации в поверхностном слое по сравнению

Адсорбция на границе раствор/воздух не поддается прямому измерению, поэтому на практике
Адсорбция на границе раствор/воздух не поддается прямому измерению, поэтому на практике
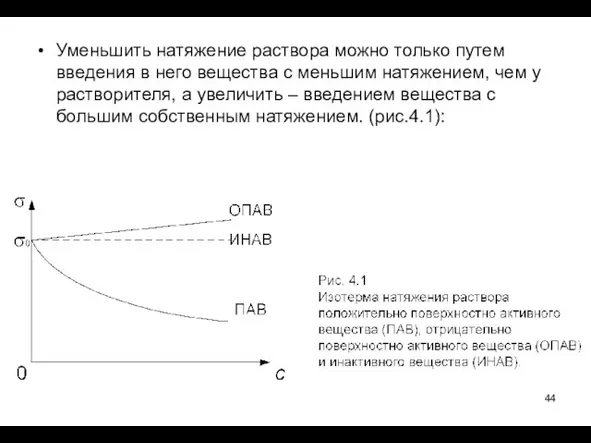
Уменьшить натяжение раствора можно только путем введения в него вещества с
Уменьшить натяжение раствора можно только путем введения в него вещества с

ПАВ – вещества, способные изменять натяжение раствора при изменении их концентрации
ПАВ – вещества, способные изменять натяжение раствора при изменении их концентрации
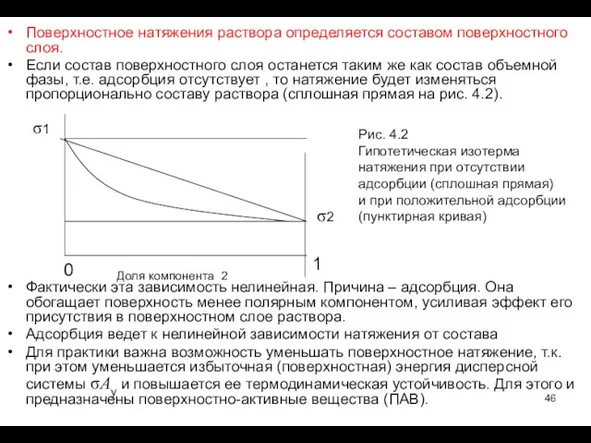
Поверхностное натяжения раствора определяется составом поверхностного слоя.
Если состав поверхностного слоя
Поверхностное натяжения раствора определяется составом поверхностного слоя.
Если состав поверхностного слоя

По химическому строению ПАВ – дифильные вещества – состоят из полярной
По химическому строению ПАВ – дифильные вещества – состоят из полярной
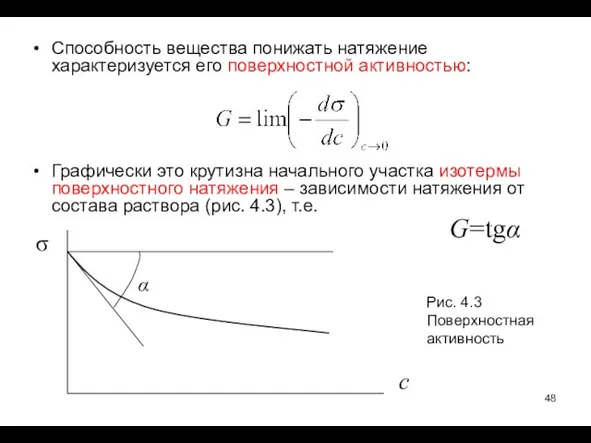
Способность вещества понижать натяжение характеризуется его поверхностной активностью:
Графически это крутизна начального
Графически это крутизна начального

Поверхностная активность определяется соотношением сродства к воде полярной и неполярной частей
Поверхностная активность определяется соотношением сродства к воде полярной и неполярной частей
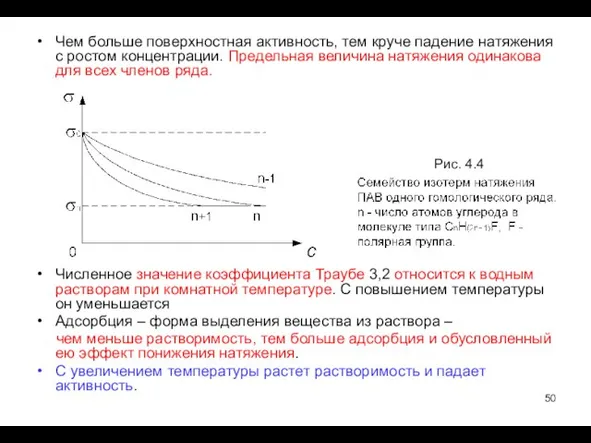
Чем больше поверхностная активность, тем круче падение натяжения с ростом концентрации.
Чем больше поверхностная активность, тем круче падение натяжения с ростом концентрации.

Эмпирическая формула Шишковского:
Аналитическое описание изотермы натяжения
Δσ = σo- σ =Β ln
Эмпирическая формула Шишковского:
Аналитическое описание изотермы натяжения
Δσ = σo- σ =Β ln

Изотерма адсорбции. Анализ уравнения Гиббса
Рассмотренная выше на качественном уровне связь адсорбции
Изотерма адсорбции. Анализ уравнения Гиббса
Рассмотренная выше на качественном уровне связь адсорбции

Однако, в соответствии с уравнением Гиббса (3.1) – при большой концентрации
Однако, в соответствии с уравнением Гиббса (3.1) – при большой концентрации
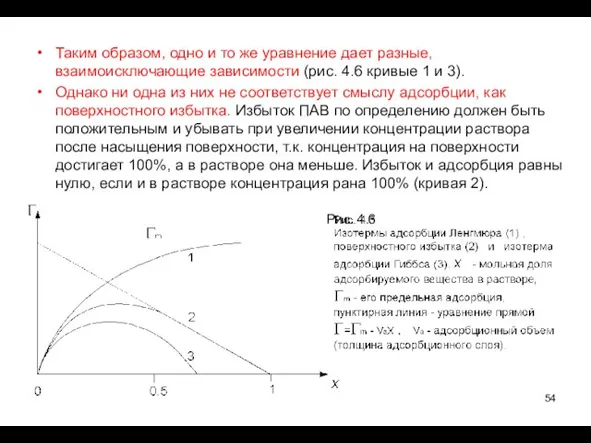
Таким образом, одно и то же уравнение дает разные, взаимоисключающие зависимости
Таким образом, одно и то же уравнение дает разные, взаимоисключающие зависимости

Причина несовпадения изотерм адсорбции (графиков 1 и 3), полученных разными способами
Причина несовпадения изотерм адсорбции (графиков 1 и 3), полученных разными способами
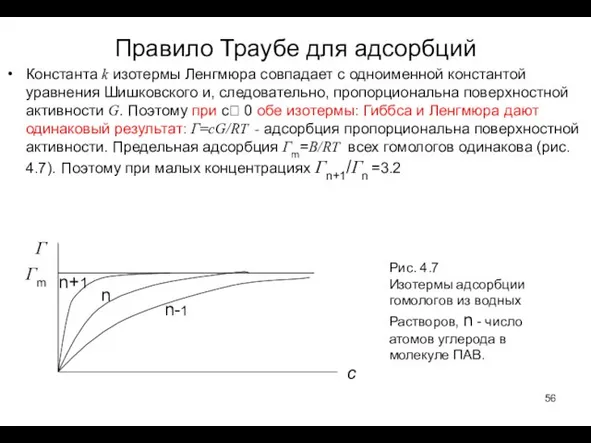
Правило Траубе для адсорбций
Константа k изотермы Ленгмюра совпадает с одноименной константой
Правило Траубе для адсорбций
Константа k изотермы Ленгмюра совпадает с одноименной константой

Полуколлоиды
Полуколлоиды – вещества способные в зависимости от условий образовывать истинные или
Полуколлоиды
Полуколлоиды – вещества способные в зависимости от условий образовывать истинные или
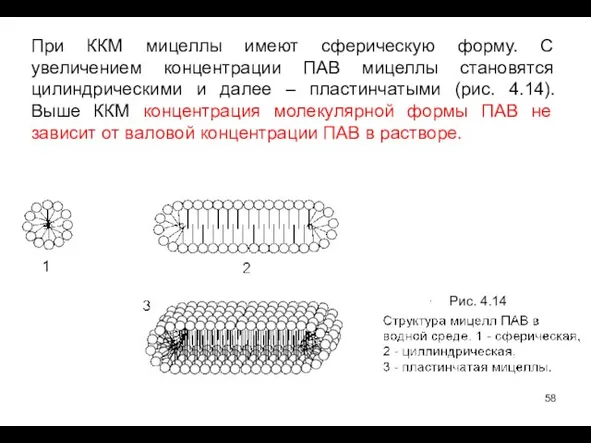
При ККМ мицеллы имеют сферическую форму. С увеличением концентрации ПАВ мицеллы
При ККМ мицеллы имеют сферическую форму. С увеличением концентрации ПАВ мицеллы

Применение мицеллярных растворов
Солюбилизация – растворение нерастворимого. Мицеллярные растворы способны растворять вещества,
Применение мицеллярных растворов
Солюбилизация – растворение нерастворимого. Мицеллярные растворы способны растворять вещества,

Строение адсорбционных слоев
Опыты Ленгмюра
Рис. 4.15
Нерастворимое в воде ПАВ, типа стеариновой кислоты,
Строение адсорбционных слоев
Опыты Ленгмюра
Рис. 4.15
Нерастворимое в воде ПАВ, типа стеариновой кислоты,
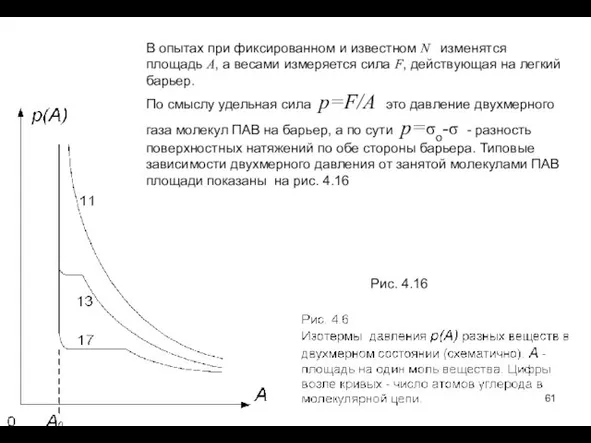
В опытах при фиксированном и известном N изменятся площадь A, а
В опытах при фиксированном и известном N изменятся площадь A, а

Большой величине площади соответствует малая величине адсорбции N/A и малая степень
Большой величине площади соответствует малая величине адсорбции N/A и малая степень

При сжатии адсорбционного слоя может наступить его конденсация – он становится
При сжатии адсорбционного слоя может наступить его конденсация – он становится

Опыты Ленгмюра впервые позволили измерить размеры молекул ПАВ – посадочную площадку
Опыты Ленгмюра впервые позволили измерить размеры молекул ПАВ – посадочную площадку

Капиллярные явления
Капиллярные явления обусловлены кривизной границы раздела двух фаз. Следствие этих
Капиллярные явления
Капиллярные явления обусловлены кривизной границы раздела двух фаз. Следствие этих
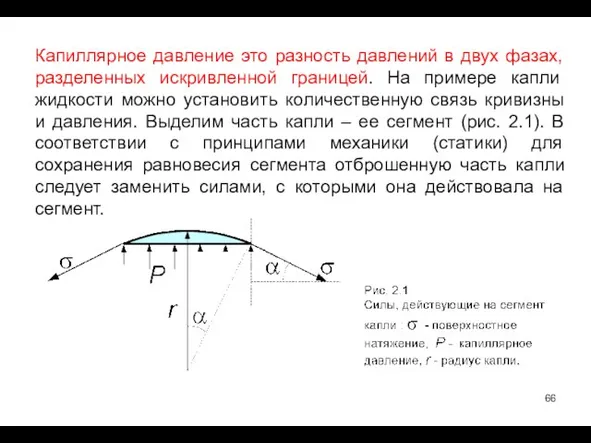
Капиллярное давление это разность давлений в двух фазах, разделенных искривленной границей.
Капиллярное давление это разность давлений в двух фазах, разделенных искривленной границей.

Это сила поверхностного натяжения, пропорциональная длине окружности основания сегмента и сила
Это сила поверхностного натяжения, пропорциональная длине окружности основания сегмента и сила

Обобщение понятие радиуса в формуле Лапласа на случай несферической капли
При
Обобщение понятие радиуса в формуле Лапласа на случай несферической капли
При

В равновесии давление в разных местах внутри замкнутой поверхности (и сосуда)
В равновесии давление в разных местах внутри замкнутой поверхности (и сосуда)

Твердым веществам анизотропия присуща в силу их кристаллического строения, поэтому различные
Твердым веществам анизотропия присуща в силу их кристаллического строения, поэтому различные

Сплавы, керамика и другие кристаллические материалы - это поликристаллические вещества. В
Сплавы, керамика и другие кристаллические материалы - это поликристаллические вещества. В

2.2 Капиллярные явления в трехфазных системах
Пример трехфазной системы - капля
2.2 Капиллярные явления в трехфазных системах
Пример трехфазной системы - капля

Общая линия соприкосновения трех фаз называется периметром смачивания. На схеме (рис.
Общая линия соприкосновения трех фаз называется периметром смачивания. На схеме (рис.

Равновесная форма капли характеризуется величиной угла Θ. Он называется краевым углом
Равновесная форма капли характеризуется величиной угла Θ. Он называется краевым углом

Вещества, которые смачиваются водой, называются гидрофильными. Это вещества с ионным типом
Вещества, которые смачиваются водой, называются гидрофильными. Это вещества с ионным типом

Влияние кривизны поверхности на свойства веществ
Переход одной из фаз гетерогенной системы
Влияние кривизны поверхности на свойства веществ
Переход одной из фаз гетерогенной системы

При равновесии на туже величину меняется потенциал и в газовой фазе.
При равновесии на туже величину меняется потенциал и в газовой фазе.

Капиллярное поднятие
Обозначения: σ=σ23 , rk – радиус капилляра,
r – радиус кривизны
Капиллярное поднятие
Обозначения: σ=σ23 , rk – радиус капилляра,
r – радиус кривизны
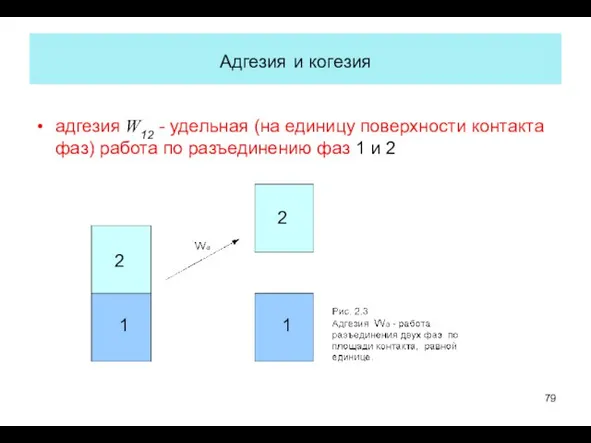
Адгезия и когезия
адгезия W12 - удельная (на единицу поверхности контакта фаз)
Адгезия и когезия
адгезия W12 - удельная (на единицу поверхности контакта фаз)

Из определения натяжения как работы образования межфазной поверхности непосредственно следует:
W12 =
Из определения натяжения как работы образования межфазной поверхности непосредственно следует:
W12 =

При полном смачивании (Θ = 0 , cosΘ = 1 )

Адсорбция ПАВ на поверхности твердых веществ
Все упомянутые выше результаты и зависимости,
Адсорбция ПАВ на поверхности твердых веществ
Все упомянутые выше результаты и зависимости,

Из водных растворов адсорбция идет на гидрофобных адсорбентах (сажа, графит, парафин,
Из водных растворов адсорбция идет на гидрофобных адсорбентах (сажа, графит, парафин,

Хемосорбция
Хемосорбция – это адсорбция за счет химического взаимодействия полярной группы с
Хемосорбция
Хемосорбция – это адсорбция за счет химического взаимодействия полярной группы с

Адсорбция в металлах и сплавах
Металлы и их сплавы – типичные представители
Адсорбция в металлах и сплавах
Металлы и их сплавы – типичные представители

Тепловые эффекты и теплоты адсорбции и смачивания
Адсорбенты – твердые вещества с
Тепловые эффекты и теплоты адсорбции и смачивания
Адсорбенты – твердые вещества с

Тепловые эффекты
Q
dQ/dΓ
Γ, m
Рис. 4.11
Интегральный Q и дифференциальный dQ/dΓ тепловой эффект
Тепловые эффекты
Q
dQ/dΓ
Γ, m
Рис. 4.11
Интегральный Q и дифференциальный dQ/dΓ тепловой эффект
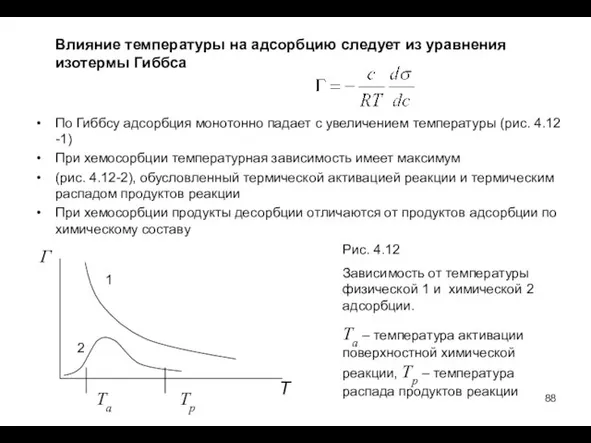
Влияние температуры на адсорбцию следует из уравнения изотермы Гиббса
По Гиббсу адсорбция
Влияние температуры на адсорбцию следует из уравнения изотермы Гиббса
По Гиббсу адсорбция

Двойной электрический слой
(ДЭС)
Двойной электрический слой
(ДЭС)

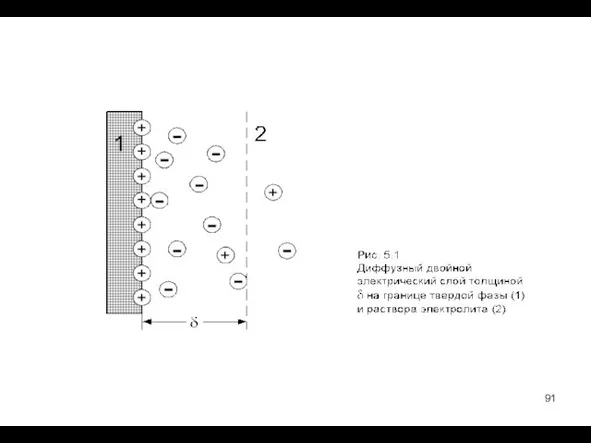
Образование и строение ДЭС
ДЭС – система пространственно разделенных эквивалентных зарядов противоположного
Образование и строение ДЭС
ДЭС – система пространственно разделенных эквивалентных зарядов противоположного

Правило избирательной адсорбции ионов
(правило Фаянса)
Избирательно адсорбируются и заряжают поверхность те ионы,
Правило избирательной адсорбции ионов
(правило Фаянса)
Избирательно адсорбируются и заряжают поверхность те ионы,

Иначе говоря, знак и величина заряда поверхности определяется в этих особых
Иначе говоря, знак и величина заряда поверхности определяется в этих особых

Из-за растворимости твердой фазы или диссоциации воды в растворе всегда присутствуют
Из-за растворимости твердой фазы или диссоциации воды в растворе всегда присутствуют

Теория диффузного ДЭС
Теория описывает строение внешней части ДЭС. Состояние внутренней части
Теория диффузного ДЭС
Теория описывает строение внешней части ДЭС. Состояние внутренней части

Количественно действие электрического поля характеризуется потенциальной энергией катионов z+FΨ и анионов

Во внешней части ДЭС объемная плотность заряда ρ отлична от нуля
Во внешней части ДЭС объемная плотность заряда ρ отлична от нуля

Распределение в пространстве заряда и потенциала подчиняется фундаментальному закону электростатики
Распределение в пространстве заряда и потенциала подчиняется фундаментальному закону электростатики

Другая форма левой части уравнения (5.7) d(dΨ/dx)/dx.
Так что после умножения
Другая форма левой части уравнения (5.7) d(dΨ/dx)/dx.
Так что после умножения
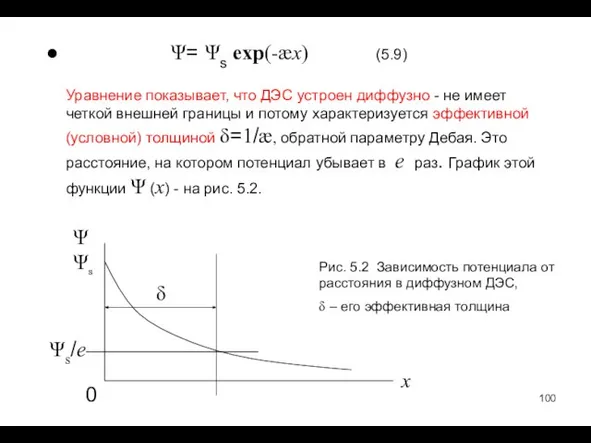
Ψ= Ψs exp(-æx) (5.9)
Уравнение показывает, что ДЭС устроен диффузно - не
Ψ= Ψs exp(-æx) (5.9)
Уравнение показывает, что ДЭС устроен диффузно - не

Интегрирование уравнения Пуассона дает заряд qv внешней части ДЭС
(5.10)
По
Интегрирование уравнения Пуассона дает заряд qv внешней части ДЭС
(5.10)
По

Развитие теории ДЭС
Электрическая емкость ДЭС доступна прямому измерению – это емкость
Развитие теории ДЭС
Электрическая емкость ДЭС доступна прямому измерению – это емкость

Строение ДЭС с учетом размера ионов
Рис. 5.3
Строение ДЭС с учетом размера ионов
Рис. 5.3
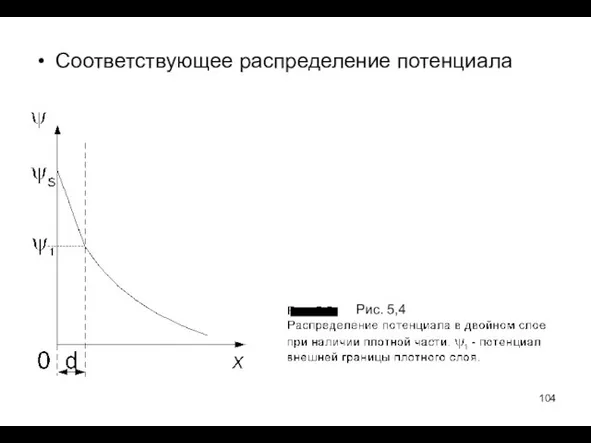
Соответствующее распределение потенциала
Рис. 5,4
Соответствующее распределение потенциала
Рис. 5,4

Фактически показанное на рис. (5.3 и 5.4) распределение ионов и потенциала
Фактически показанное на рис. (5.3 и 5.4) распределение ионов и потенциала

Влияние электролитов на строение ДЭС
В рамках изложенных представлений ДЭС исчерпывающе характеризуется
Влияние электролитов на строение ДЭС
В рамках изложенных представлений ДЭС исчерпывающе характеризуется
Распределение потенциала
Ψ
Ψs
ζ1
ζ2
ζ3=0
x
c1
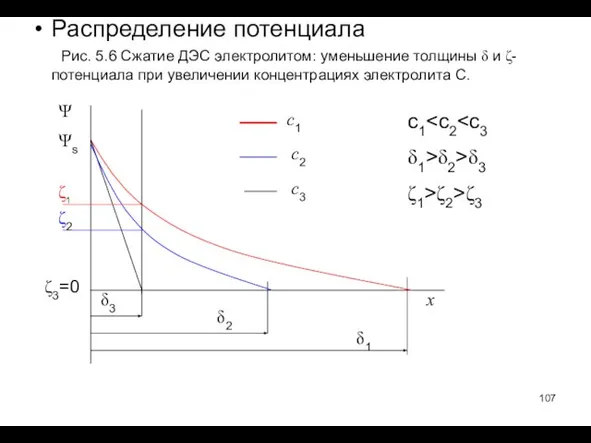
Рис. 5.6 Сжатие ДЭС электролитом: уменьшение толщины δ и
Распределение потенциала
Ψ
Ψs
ζ1
ζ2
ζ3=0
x
c1
Рис. 5.6 Сжатие ДЭС электролитом: уменьшение толщины δ и

Влияние электролитов на ζ-потенциал
Сжатие ДЭС электролитом вызывает уменьшение ζ-потенциала, вплоть до
Влияние электролитов на ζ-потенциал
Сжатие ДЭС электролитом вызывает уменьшение ζ-потенциала, вплоть до

Действие ПО электролитов на ДЭС
Представленный выше вариант теории, как и более
Действие ПО электролитов на ДЭС
Представленный выше вариант теории, как и более

Электрокапиллярность
Электрокапиллярные эффекты – зависимость поверхностного натяжения межфазной границы от разности электрических
Электрокапиллярность
Электрокапиллярные эффекты – зависимость поверхностного натяжения межфазной границы от разности электрических
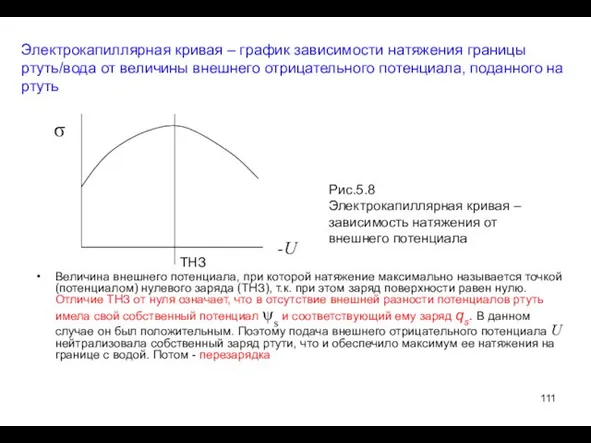
Электрокапиллярная кривая – график зависимости натяжения границы ртуть/вода от величины внешнего
Электрокапиллярная кривая – график зависимости натяжения границы ртуть/вода от величины внешнего

Уравнение электрокапиллярной кривой – уравнение Липпмана:
qs= - dσ/dU (5.15)
На левой ветви
Уравнение электрокапиллярной кривой – уравнение Липпмана:
qs= - dσ/dU (5.15)
На левой ветви

В формуле (5.16) присутствуют заряд и потенциал. Они могут быть связаны
В формуле (5.16) присутствуют заряд и потенциал. Они могут быть связаны
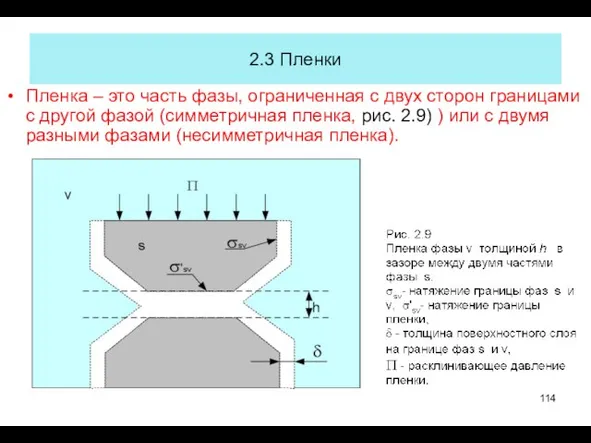
2.3 Пленки
Пленка – это часть фазы, ограниченная с двух сторон границами
2.3 Пленки
Пленка – это часть фазы, ограниченная с двух сторон границами

Пленки делятся на толстые и тонкие. Толстая пленка имеет толщину h
Пленки делятся на толстые и тонкие. Толстая пленка имеет толщину h

Пленка, которая стремится к самопроизвольному увеличению толщины создает положительное расклинивающее давление
Пленка, которая стремится к самопроизвольному увеличению толщины создает положительное расклинивающее давление

Сближение тел, ограничивающих толщину пленки, требует совершения работы w против силы
Сближение тел, ограничивающих толщину пленки, требует совершения работы w против силы

Устойчивость дисперсных систем
Коллоидные растворы (и другие дисперсные системы) термодинамически неустойчивы –
Устойчивость дисперсных систем
Коллоидные растворы (и другие дисперсные системы) термодинамически неустойчивы –

Если таких мер не предпринимать, то во взаимодействии частиц будет преобладать
Если таких мер не предпринимать, то во взаимодействии частиц будет преобладать

В вакууме, и в газовой среде молекулярные силы всегда ведут к
В вакууме, и в газовой среде молекулярные силы всегда ведут к

Иначе говоря, жидкая дисперсионная среда, обладающая хорошим сродством к поверхности частиц
Иначе говоря, жидкая дисперсионная среда, обладающая хорошим сродством к поверхности частиц

Отталкивание может иметь разную природу.
Природа сил отталкивания (факторы агрегативной устойчивости): электростатическая
Отталкивание может иметь разную природу.
Природа сил отталкивания (факторы агрегативной устойчивости): электростатическая

Электростатическое отталкивание ДЭС
(Электростатическая составляющая расклинивающего давления)
Между двумя телами, разделенными пленкой жидкости
Электростатическое отталкивание ДЭС
(Электростатическая составляющая расклинивающего давления)
Между двумя телами, разделенными пленкой жидкости
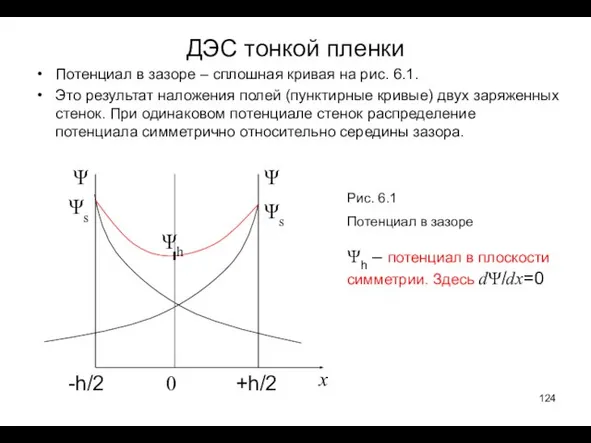
ДЭС тонкой пленки
Потенциал в зазоре – сплошная кривая на рис. 6.1.
ДЭС тонкой пленки
Потенциал в зазоре – сплошная кривая на рис. 6.1.

Распределение ионов и потенциала в зазоре подчиняется тем же законам, что
Распределение ионов и потенциала в зазоре подчиняется тем же законам, что

После звлечения корня квадратного имеем:
dΨ/dx= æ (Ψ2 - Ψh2)1/2.
Интегрируя это
После звлечения корня квадратного имеем:
dΨ/dx= æ (Ψ2 - Ψh2)1/2.
Интегрируя это

В формуле (6.2) потенциал Ψh нужно заменить не зависящим от h
В формуле (6.2) потенциал Ψh нужно заменить не зависящим от h

Производная функции (6.3): dΨ/dx = æΨssh(æx) / ch(æh/2).
При x= - h/2
Производная функции (6.3): dΨ/dx = æΨssh(æx) / ch(æh/2).
При x= - h/2

Молекулярное притяжение
Расчет энергии молекулярного притяжения двух плоских тел, разделенных узким зазором
Молекулярное притяжение
Расчет энергии молекулярного притяжения двух плоских тел, разделенных узким зазором
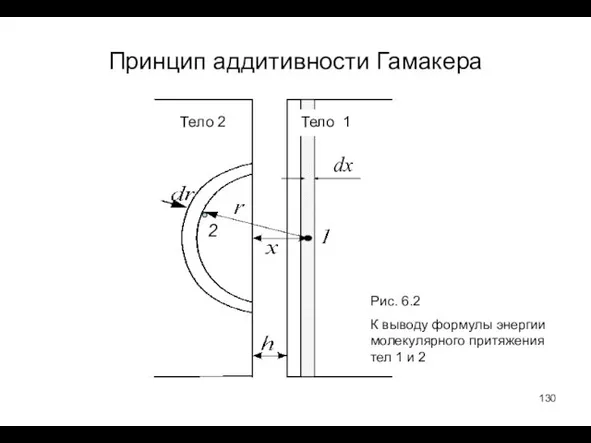
Принцип аддитивности Гамакера
Тело 2
Рис. 6.2
К выводу формулы энергии молекулярного притяжения тел
Принцип аддитивности Гамакера
Тело 2
Рис. 6.2
К выводу формулы энергии молекулярного притяжения тел

Выберем (рис. 6.2) две произвольные молекулы 1 в теле 1 и
Выберем (рис. 6.2) две произвольные молекулы 1 в теле 1 и

Все тело 2 представимо как совокупность сферических слоев (половины качана капусты).
Все тело 2 представимо как совокупность сферических слоев (половины качана капусты).

Тело 1 представимо как совокупность тонких плоских слоев толщиной dx. Объем
Тело 1 представимо как совокупность тонких плоских слоев толщиной dx. Объем

Та же формула в компактной записи
Uм= - AH / h2 (6.11)
AH=
Та же формула в компактной записи
Uм= - AH / h2 (6.11)
AH=

Теория взаимодействия фаз Лифшица
По Лифшицу тела взаимодействуют между собой своими электромагнитными
Теория взаимодействия фаз Лифшица
По Лифшицу тела взаимодействуют между собой своими электромагнитными

Динамические поля тел, в отличие от статического поля ДЭС, создают силы
Динамические поля тел, в отличие от статического поля ДЭС, создают силы

Выполнение соответствующих расчетов требует задания набора частот, амплитуд и фаз электромагнитных
Выполнение соответствующих расчетов требует задания набора частот, амплитуд и фаз электромагнитных

Учет эффекта электромагнитного запаздывания ведет к тому, что теория Лифшица дает
Учет эффекта электромагнитного запаздывания ведет к тому, что теория Лифшица дает

Теория ДЛФО
Теория ДЛФО

Переход Дерягина
Для объяснения свойств коллоидных растворов необходимо анализировать взаимодействие сферических частиц,
Переход Дерягина
Для объяснения свойств коллоидных растворов необходимо анализировать взаимодействие сферических частиц,
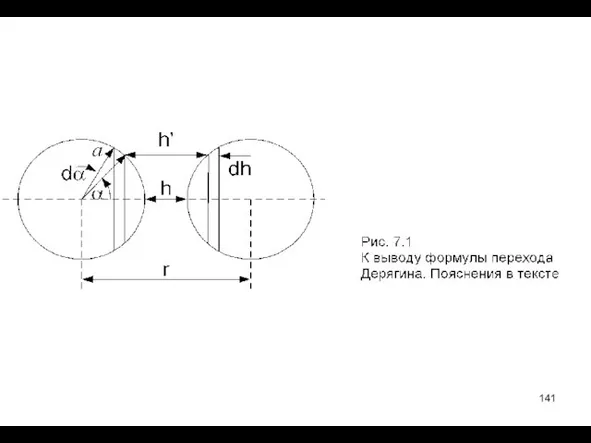

На поверхности сфер радиуса a выделим симметричные пояски шириной dh. Положим,
На поверхности сфер радиуса a выделим симметричные пояски шириной dh. Положим,

Подставляя в (7.1) выражение (6.6) для энергии отталкивания плоских заряженных тел,
Подставляя в (7.1) выражение (6.6) для энергии отталкивания плоских заряженных тел,

При произвольной величине потенциала Ψs и соблюдении всех других ограничений (æh
При произвольной величине потенциала Ψs и соблюдении всех других ограничений (æh

При неограниченном увеличении потенциала функция γ (7.5) стремится к единице. Таким
При неограниченном увеличении потенциала функция γ (7.5) стремится к единице. Таким

Потенциальные кривые и устойчивость
В теории ДЛФО состояние и поведение дисперсной системы
Потенциальные кривые и устойчивость
В теории ДЛФО состояние и поведение дисперсной системы
В общем случае потенциальная кривая (рис. 7.3) имеет максимум ΔU (потенциальный
В общем случае потенциальная кривая (рис. 7.3) имеет максимум ΔU (потенциальный

Энергия взаимодействия частиц сложным образом зависит от расстояния между ними: на
Энергия взаимодействия частиц сложным образом зависит от расстояния между ними: на
Влияние электролитов на потенциальные кривые и устойчивость дисперсных систем
Влияние индифферентных электролитов
Влияние электролитов на потенциальные кривые и устойчивость дисперсных систем
Влияние индифферентных электролитов
При увеличении концентрации электролита уменьшаются силы отталкивания, поэтому уменьшается барьер, появляется
При увеличении концентрации электролита уменьшаются силы отталкивания, поэтому уменьшается барьер, появляется

В коллоидных системах, как и в молекулярных, изменение состояния системы осуществляется
В коллоидных системах, как и в молекулярных, изменение состояния системы осуществляется
![1. ΔU >> kBT, [Umin] При таком соотношении экстремумов функции взаимодействия](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/526957/slide-151.jpg)
1. ΔU >> kBT, [Umin] << kBT. - имеется большой барьер
1. ΔU >> kBT, [Umin] << kBT. - имеется большой барьер
![2. ΔU ≥ kBT, [Umin] При сопоставимости величин энергии столкновений и](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/526957/slide-152.jpg)
2. ΔU ≥ kBT, [Umin] << kBT. - барьер сопоставим по
2. ΔU ≥ kBT, [Umin] << kBT. - барьер сопоставим по
![3. ΔU >> kBT, [Umin] ≥ kBT. - большой барьер и](/_ipx/f_webp&q_80&fit_contain&s_1440x1080/imagesDir/jpg/526957/slide-153.jpg)
3. ΔU >> kBT, [Umin] ≥ kBT. - большой барьер и
3. ΔU >> kBT, [Umin] ≥ kBT. - большой барьер и
1
2
Рис. 7.6 Контактная 1 и неконтактная 2 (безбарьерная) коагуляция частиц
1
2
Рис. 7.6 Контактная 1 и неконтактная 2 (безбарьерная) коагуляция частиц

4. ΔU = 0 - потенциальный барьер равен нулю.
При таком типе
4. ΔU = 0 - потенциальный барьер равен нулю.
При таком типе

Влияние неиндифферентных (потенциалопределяющих) электролитов (см. ДЭС) связано с их действием на
Влияние неиндифферентных (потенциалопределяющих) электролитов (см. ДЭС) связано с их действием на
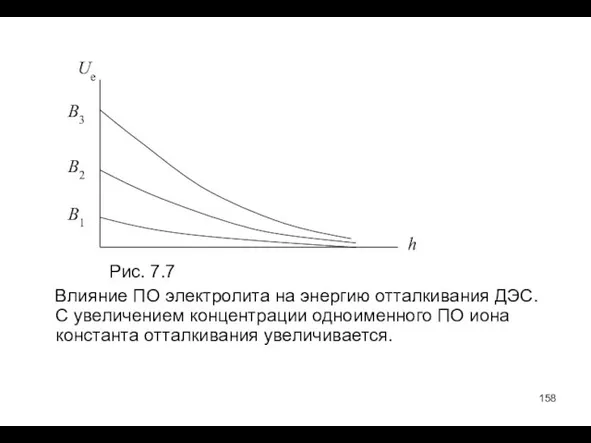
Рис. 7.7
Влияние ПО электролита на энергию отталкивания ДЭС. С увеличением
Рис. 7.7
Влияние ПО электролита на энергию отталкивания ДЭС. С увеличением

Изменение сил отталкивания под действием ПО электролита в общем вызывает такие
Изменение сил отталкивания под действием ПО электролита в общем вызывает такие

Критическая концентрация электролита и правила электролитной коагуляции
Условие быстрой необратимой коагуляции ΔU=0
Критическая концентрация электролита и правила электролитной коагуляции
Условие быстрой необратимой коагуляции ΔU=0

Из структуры формул первой и второй строки следует, что эти обе
Из структуры формул первой и второй строки следует, что эти обе

Частные виды формулы (7,12)
При большом потенциале поверхности, согласно (7.6) с точностью
Частные виды формулы (7,12)
При большом потенциале поверхности, согласно (7.6) с точностью

Закон (7.13) действует только при концентрационной коагуляции индифферентными электролитами.
Закон (7.14) действует
Закон (7.13) действует только при концентрационной коагуляции индифферентными электролитами.
Закон (7.14) действует

Эффект смещения плоскости локализации заряда относительно межфазной границы
Не существует физически обоснованных
Эффект смещения плоскости локализации заряда относительно межфазной границы
Не существует физически обоснованных

Правила электролитной коагуляции, сопоставление с теорией ДЛФО
Правила коагуляции это результат обобщение
Правила электролитной коагуляции, сопоставление с теорией ДЛФО
Правила коагуляции это результат обобщение

3 – правило альтернативное правилу 2. Коагуляция наступает при уменьшении абсолютной
3 – правило альтернативное правилу 2. Коагуляция наступает при уменьшении абсолютной

Первое правило объясняется тем, что любой электролит при возрастании концентрации увеличивает
Первое правило объясняется тем, что любой электролит при возрастании концентрации увеличивает

Получение дисперсных систем
Получение дисперсной системы сводится к решению двух задач: Получению
Получение дисперсных систем
Получение дисперсной системы сводится к решению двух задач: Получению

При химической конденсации (образование частиц AgI в растворе, дыма при горении
При химической конденсации (образование частиц AgI в растворе, дыма при горении

Критический зародыш – минимальный по размеру зародыш новой фазы, который при
Критический зародыш – минимальный по размеру зародыш новой фазы, который при

Химическая конденсация
Правила образования коллоидного раствора непосредственно при проведении химической реакции:
1. Реакция
Химическая конденсация
Правила образования коллоидного раствора непосредственно при проведении химической реакции:
1. Реакция

Метод дробления. Эффект Ребиндера.
Дроблением получают дисперсные системы с размером частиц 10-6
Метод дробления. Эффект Ребиндера.
Дроблением получают дисперсные системы с размером частиц 10-6

Пептизация
Пептизация – получение коллоидных растворов путем разрыхления осадков и перевода взвеси
Пептизация
Пептизация – получение коллоидных растворов путем разрыхления осадков и перевода взвеси
Правило осадков
Правило пептизации (правило осадков Оствальда) является общим для коллоидных растворов
Правило осадков
Правило пептизации (правило осадков Оствальда) является общим для коллоидных растворов

С позиций теории ДЛФО пептизатор это ПО электролит. Увеличение его концентрации
С позиций теории ДЛФО пептизатор это ПО электролит. Увеличение его концентрации

Молекулярно-кинетические свойства дисперсных систем
Молекулярно-кинетические свойства дисперсных систем

Седиментация и диффузия
Молекулярно кинетические свойства и явления это явления обусловленные движением
Седиментация и диффузия
Молекулярно кинетические свойства и явления это явления обусловленные движением

Уникальность коллоидов в том, что только в них интенсивность обоих типов
Уникальность коллоидов в том, что только в них интенсивность обоих типов

Формула Эйнштейна для коэффициента диффузии:
D=kT/6πηa (9.2)
6πηa - коэффициент Стокса – сила
Формула Эйнштейна для коэффициента диффузии:
D=kT/6πηa (9.2)
6πηa - коэффициент Стокса – сила

Масса частицы m=vρ, где v =4πa3/3 – объем частицы и ρ
Масса частицы m=vρ, где v =4πa3/3 – объем частицы и ρ

Седиментационно-диффузионное равновесие
Одновременное участие частиц в оседании и диффузии приводит к установлению
Седиментационно-диффузионное равновесие
Одновременное участие частиц в оседании и диффузии приводит к установлению

Уравнение седиментационно-диффузионного равновесия
n=n0 exp( -mgh/ kBT) (9.6)
По сути это закон распределения Больцмана
Уравнение седиментационно-диффузионного равновесия
n=n0 exp( -mgh/ kBT) (9.6)
По сути это закон распределения Больцмана

Нормирование распределения Больцмана
Обычно известна средняя по высоте (рецептурная) концентрация дисперсной фазы
Нормирование распределения Больцмана
Обычно известна средняя по высоте (рецептурная) концентрация дисперсной фазы

Формула (9.7) выражает условие нормирования распределения Больцмана - нахождения неизвестной константы
Формула (9.7) выражает условие нормирования распределения Больцмана - нахождения неизвестной константы
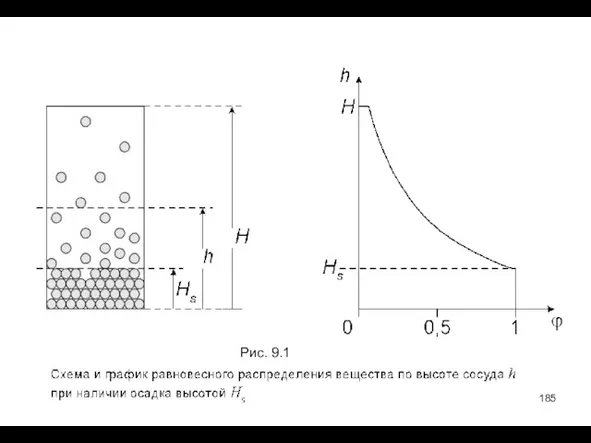
Рис. 9.1
Рис. 9.1

Высота осадка находится из условия сохранения вещества при разделении системы на
Высота осадка находится из условия сохранения вещества при разделении системы на

Электрокинетические явления
Это явления связанные со взаимным смещением фаз дисперсной системы.
Прямые электрокинетические
Электрокинетические явления
Это явления связанные со взаимным смещением фаз дисперсной системы.
Прямые электрокинетические

Электрофорез и потенциал оседания возникают в относительно разбавленных дисперсных системах, где
Электрофорез и потенциал оседания возникают в относительно разбавленных дисперсных системах, где
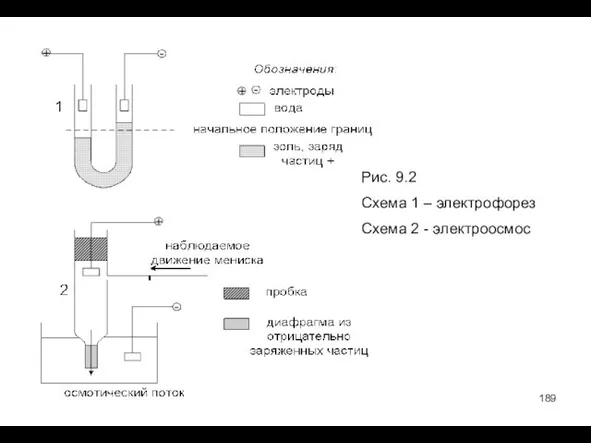
Рис. 9.2
Схема 1 – электрофорез
Схема 2 - электроосмос
Рис. 9.2
Схема 1 – электрофорез
Схема 2 - электроосмос

Механизм электрофореза
Не тривиальность явления электрофореза обусловлена тем, что в растворе электролита
Механизм электрофореза
Не тривиальность явления электрофореза обусловлена тем, что в растворе электролита
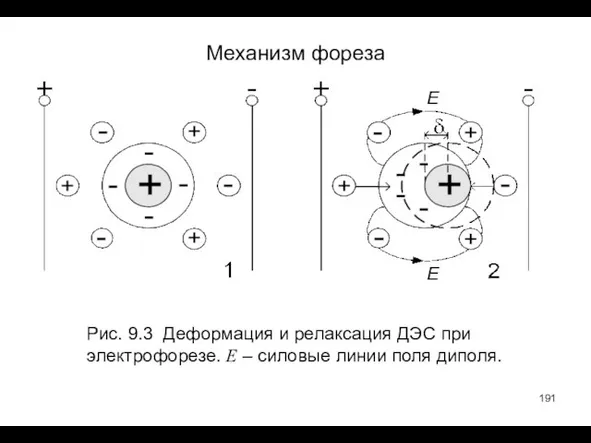
Механизм фореза
Рис. 9.3 Деформация и релаксация ДЭС при электрофорезе. E –
Механизм фореза
Рис. 9.3 Деформация и релаксация ДЭС при электрофорезе. E –

Ионы раствора притягиваются к противоположным по знаку полюсам диполя, достраивая недостающую
Ионы раствора притягиваются к противоположным по знаку полюсам диполя, достраивая недостающую
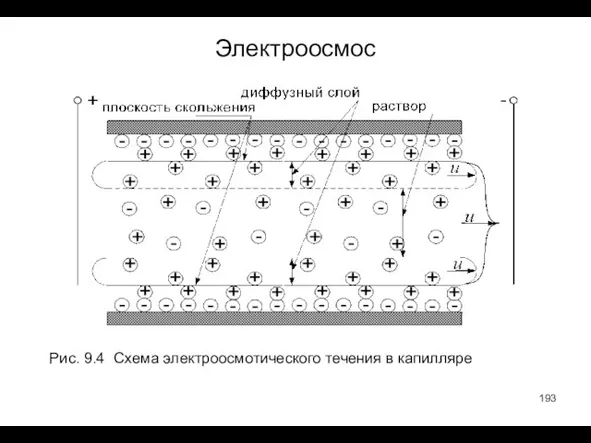
Электроосмос
Рис. 9.4 Схема электроосмотического течения в капилляре
Электроосмос
Рис. 9.4 Схема электроосмотического течения в капилляре

Пористое тело это совокупность параллельных потоку капилляров. Схема рис. 9.4 представляет
Пористое тело это совокупность параллельных потоку капилляров. Схема рис. 9.4 представляет
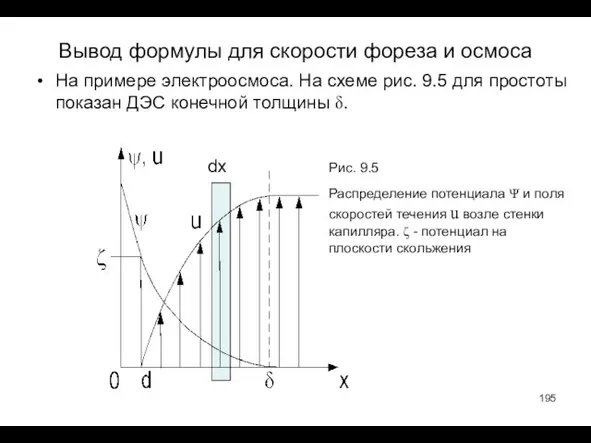
Вывод формулы для скорости фореза и осмоса
На примере электроосмоса. На схеме
Вывод формулы для скорости фореза и осмоса
На примере электроосмоса. На схеме

Силы, действующие внутри ДЭС на слой жидкости толщиной dx и площадью
Силы, действующие внутри ДЭС на слой жидкости толщиной dx и площадью

Результирующая сила трения fv равна разнице сил трения среды о внешнюю
Результирующая сила трения fv равна разнице сил трения среды о внешнюю

Второе интегрирование - (после умножения на dx) от x=d где Ψ=ζ
Второе интегрирование - (после умножения на dx) от x=d где Ψ=ζ

Объемная скорость электроосмоса v=us, где s - площадь поперечного сечения всех
Объемная скорость электроосмоса v=us, где s - площадь поперечного сечения всех
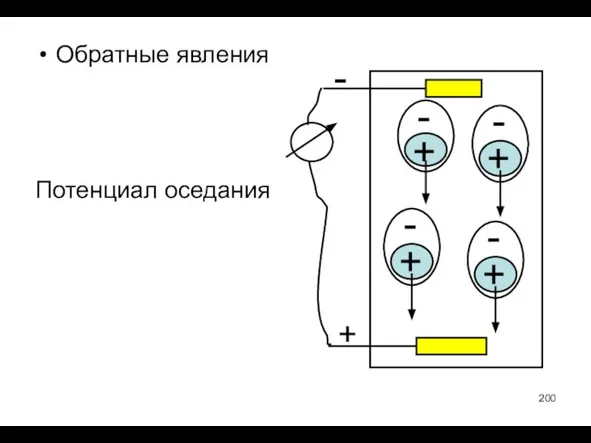
Обратные явления
Потенциал оседания
+
-
Обратные явления
Потенциал оседания
+
-
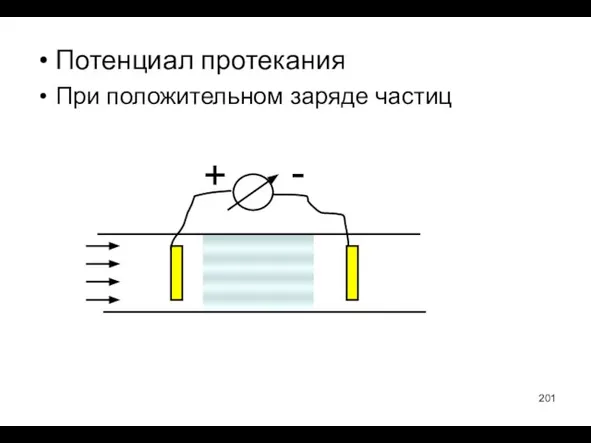
Потенциал протекания
При положительном заряде частиц
+
-
Потенциал протекания
При положительном заряде частиц
+
-

Кинетика коагуляции
Теория ДЛФО формулирует условие быстрой необратимой коагуляции, а кинетику этого
Кинетика коагуляции
Теория ДЛФО формулирует условие быстрой необратимой коагуляции, а кинетику этого

При R ≤ Rk , т.е. при столкновении, вступает в действие
При R ≤ Rk , т.е. при столкновении, вступает в действие
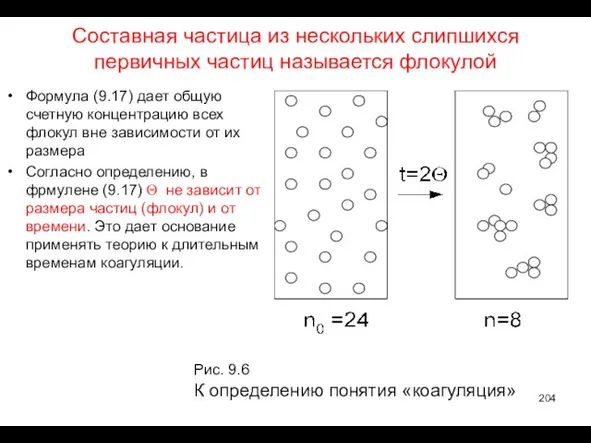
Составная частица из нескольких слипшихся первичных частиц называется флокулой
Формула (9.17) дает
Составная частица из нескольких слипшихся первичных частиц называется флокулой
Формула (9.17) дает

Удобной характеристикой текущего состояния коагулирующей системы является среднее число частиц в
Удобной характеристикой текущего состояния коагулирующей системы является среднее число частиц в

Теория Смолуховского дает и более детальное описание процесса коагуляции, а именно,
Теория Смолуховского дает и более детальное описание процесса коагуляции, а именно,

Оптические свойства дисперсных систем
Прохождение света через любую оптическую среду сопровождается его
Оптические свойства дисперсных систем
Прохождение света через любую оптическую среду сопровождается его

Соответственно, коэффициент ослабления K складывается из коэффициента поглощения Kп и коэффициента
Соответственно, коэффициент ослабления K складывается из коэффициента поглощения Kп и коэффициента

Электрическое поле световой волны индуцирует в частицах дипольный момент, который осциллирует
Электрическое поле световой волны индуцирует в частицах дипольный момент, который осциллирует
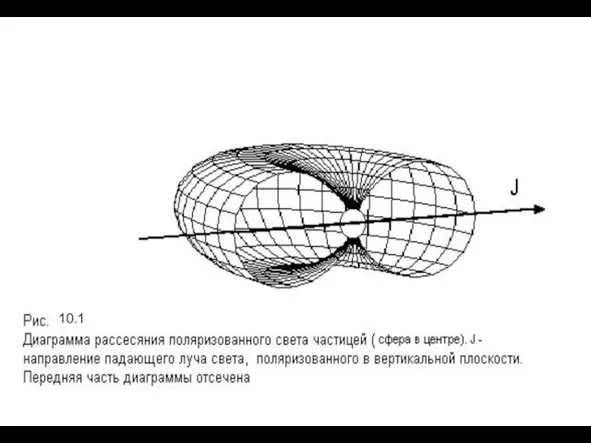
10.1
10.1

Диаграммы рассеяния естественного света получается суперпозицией (суммой) множества диаграмм, получаемых вращением
Диаграммы рассеяния естественного света получается суперпозицией (суммой) множества диаграмм, получаемых вращением
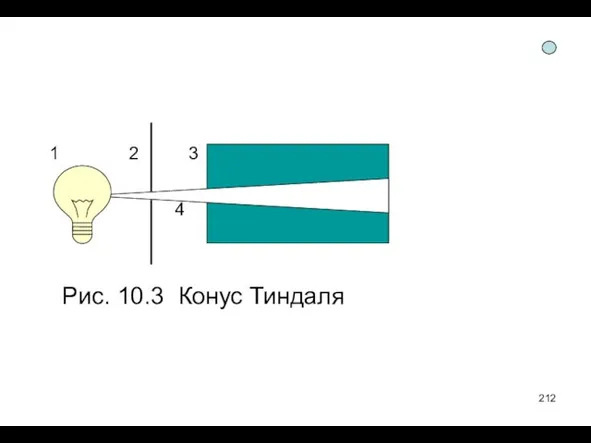
Рис. 10.3 Конус Тиндаля
1
2
3
4
Рис. 10.3 Конус Тиндаля
1
2
3
4

Структура и структурирование дисперсных систем
Структура – характеристика взаимного расположения частиц.
Структурирование –
Структура и структурирование дисперсных систем
Структура – характеристика взаимного расположения частиц.
Структурирование –
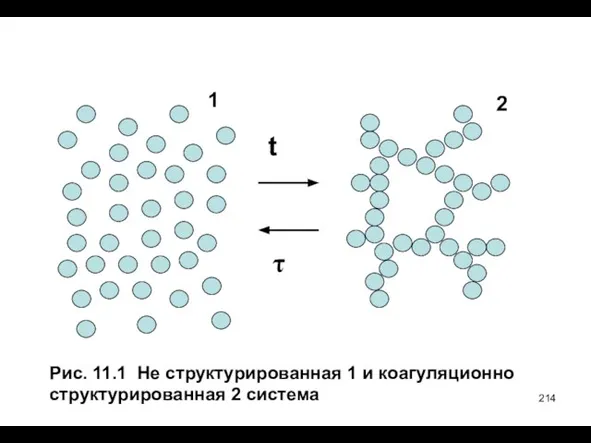
Рис. 11.1 Не структурированная 1 и коагуляционно структурированная 2 система
1
2
t
τ
Рис. 11.1 Не структурированная 1 и коагуляционно структурированная 2 система
1
2
t
τ

Наиболее распространено коагуляционное структурирование, обычно при обратимой безбарьерной коагуляции (ΔU>>kT, Umin>kT).
Наиболее распространено коагуляционное структурирование, обычно при обратимой безбарьерной коагуляции (ΔU>>kT, Umin>kT).

Фрактальная размерность
Рис. 11.2. Флокула
r
L
ν=(L / r)ф
ν=1+t/t*
t*=3η/8kTnн
Фрактальная размерность
Рис. 11.2. Флокула
r
L
ν=(L / r)ф
ν=1+t/t*
t*=3η/8kTnн

Фрактальная размерность (рис. 11.2) – показатель степени ф, определяющий зависимость размера
Фрактальная размерность (рис. 11.2) – показатель степени ф, определяющий зависимость размера

Минимально возможное число связанных частиц во флокуле реализуется, если они расположены
Минимально возможное число связанных частиц во флокуле реализуется, если они расположены

Коагуляционное структурирование
В процессе коагуляции среднее число частиц в одной флокуле растет
Коагуляционное структурирование
В процессе коагуляции среднее число частиц в одной флокуле растет

Крупные флокулы более рыхлые, чем мелкие и потому менее прочные. В
Крупные флокулы более рыхлые, чем мелкие и потому менее прочные. В

Структурирование в поле обусловлено дипольным взаимодействием частиц UII и UT. Оно
Структурирование в поле обусловлено дипольным взаимодействием частиц UII и UT. Оно

Структурирование устойчивых систем за счет ограниченности объема (ϕ≥0.52)
В условиях отсутствия свободного
Структурирование устойчивых систем за счет ограниченности объема (ϕ≥0.52)
В условиях отсутствия свободного

Прочность (энергия ΔU) фиксации частиц за счет отталкивания (на примере потенциальных
Прочность (энергия ΔU) фиксации частиц за счет отталкивания (на примере потенциальных

Реология
Наука о деформационных свойствах материалов
Напряжение сдвига τ =F / A, деформация
Реология
Наука о деформационных свойствах материалов
Напряжение сдвига τ =F / A, деформация
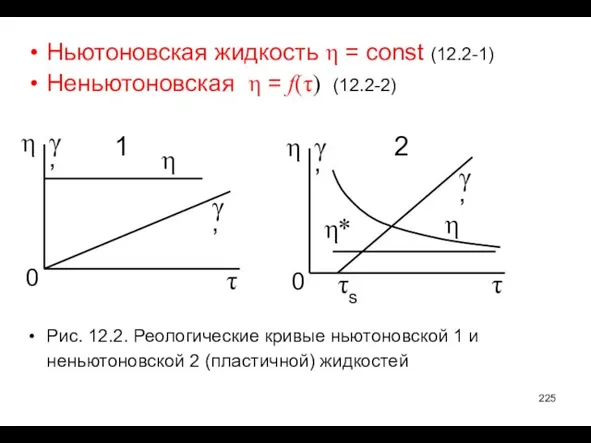
Ньютоновская жидкость η = const (12.2-1)
Неньютоновская η = f(τ) (12.2-2)
Рис. 12.2.
Ньютоновская жидкость η = const (12.2-1)
Неньютоновская η = f(τ) (12.2-2)
Рис. 12.2.

Пластичность
ПЛАСТИЧНОСТЬ – способность к неограниченным по величине деформациям без разрушения материала.
Пластичность
ПЛАСТИЧНОСТЬ – способность к неограниченным по величине деформациям без разрушения материала.

Принадлежность к тому или иному типу определяется структурой системы
Ньютоновские – неструктурированные
Вязкость
Принадлежность к тому или иному типу определяется структурой системы
Ньютоновские – неструктурированные
Вязкость

Реология тиксотропных систем
Применение формул Эйнштейна-Бринкмена к флокулированным системам
η=η0 / (1- ϕe)α (12.6)
ϕe=1
Реология тиксотропных систем
Применение формул Эйнштейна-Бринкмена к флокулированным системам
η=η0 / (1- ϕe)α (12.6)
ϕe=1
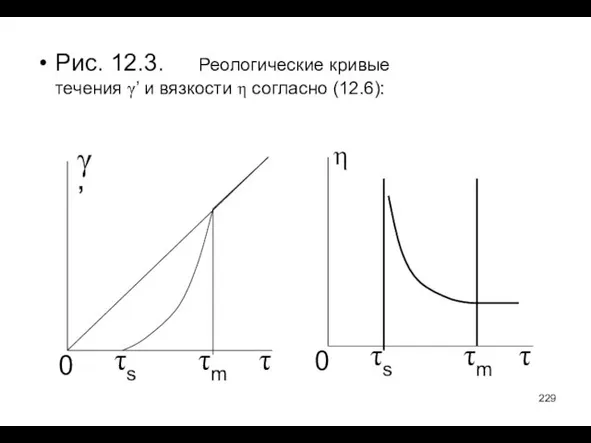
Рис. 12.3. Реологические кривые течения γ’ и вязкости η согласно (12.6):
γ’
τ
τs
τm
0
Рис. 12.3. Реологические кривые течения γ’ и вязкости η согласно (12.6):
γ’
τ
τs
τm
0
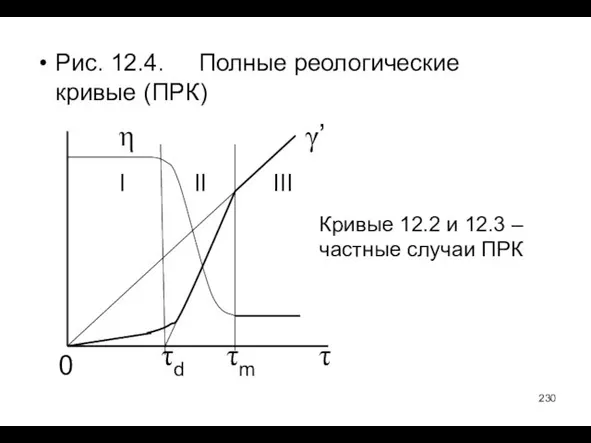
Рис. 12.4. Полные реологические кривые (ПРК)
I
II
III
γ’
η
0
τd
τm
τ
Кривые 12.2 и 12.3 – частные
Рис. 12.4. Полные реологические кривые (ПРК)
I
II
III
γ’
η
0
τd
τm
τ
Кривые 12.2 и 12.3 – частные

Реология ПКС
Рис. 12.5.
Течение ПКС (слайд 222) без увеличя объема системы (дилатации)
Реология ПКС
Рис. 12.5.
Течение ПКС (слайд 222) без увеличя объема системы (дилатации)
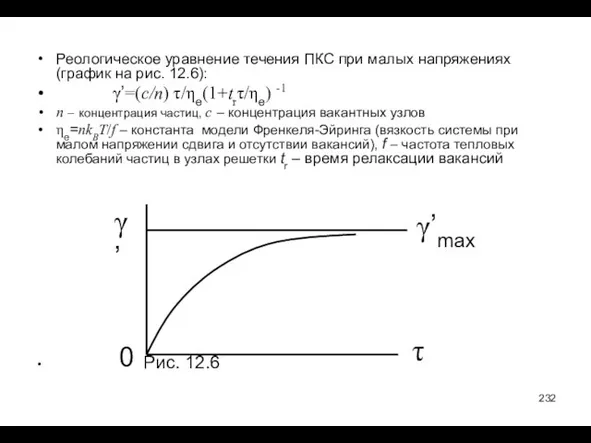
Реологическое уравнение течения ПКС при малых напряжениях (график на рис. 12.6):
Реологическое уравнение течения ПКС при малых напряжениях (график на рис. 12.6):
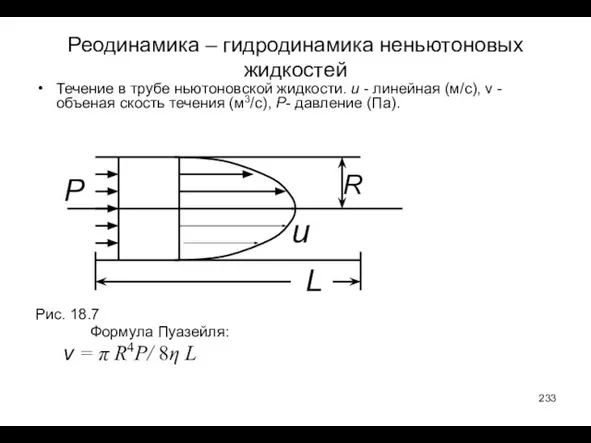
Реодинамика – гидродинамика неньютоновых жидкостей
Течение в трубе ньютоновской жидкости. u -
Реодинамика – гидродинамика неньютоновых жидкостей
Течение в трубе ньютоновской жидкости. u -
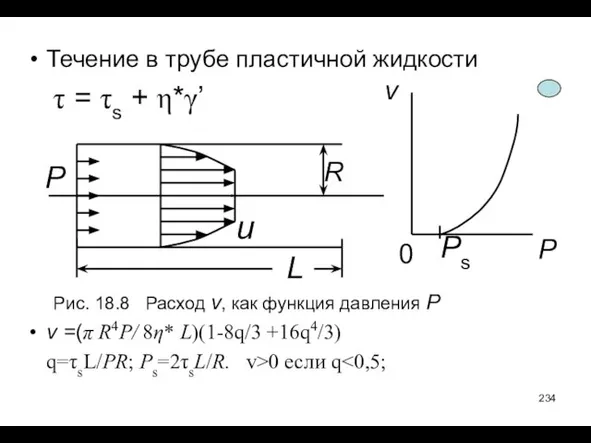
Течение в трубе пластичной жидкости
τ = τs + η*γ’
Рис. 18.8 Расход
Течение в трубе пластичной жидкости
τ = τs + η*γ’
Рис. 18.8 Расход
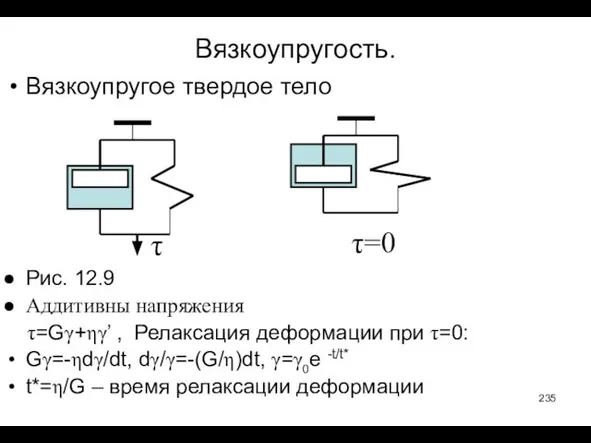
Вязкоупругость.
Вязкоупругое твердое тело
Рис. 12.9
Аддитивны напряжения
τ=Gγ+ηγ’ , Релаксация деформации при
Вязкоупругость.
Вязкоупругое твердое тело
Рис. 12.9
Аддитивны напряжения
τ=Gγ+ηγ’ , Релаксация деформации при
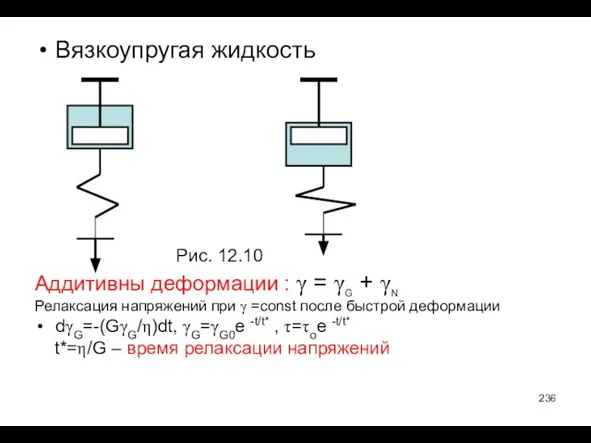
Вязкоупругая жидкость
Рис. 12.10
Аддитивны деформации : γ = γG + γN
Релаксация
Вязкоупругая жидкость
Рис. 12.10
Аддитивны деформации : γ = γG + γN
Релаксация

Дисперсные системы
Дисперсные системы
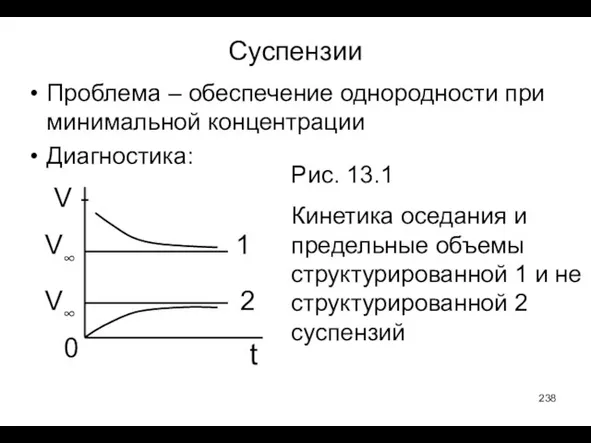
Суспензии
Проблема – обеспечение однородности при минимальной концентрации
Диагностика:
V
t
0
V∞
V∞
1
2
Рис. 13.1
Кинетика оседания и предельные
Суспензии
Проблема – обеспечение однородности при минимальной концентрации
Диагностика:
V
t
0
V∞
V∞
1
2
Рис. 13.1
Кинетика оседания и предельные
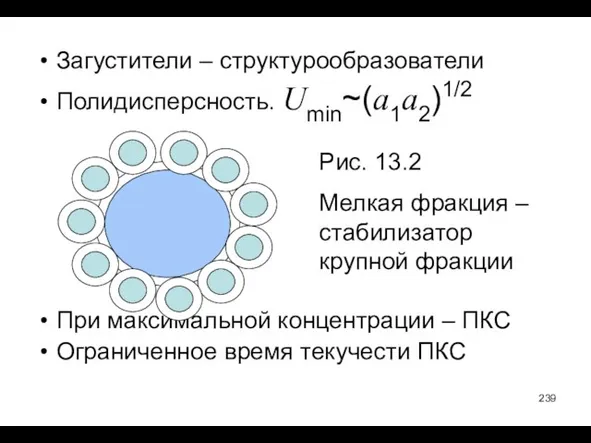
Загустители – структурообразователи
Полидисперсность. Umin~(a1a2)1/2
При максимальной концентрации – ПКС
Ограниченное время текучести ПКС
Рис.
Загустители – структурообразователи
Полидисперсность. Umin~(a1a2)1/2
При максимальной концентрации – ПКС
Ограниченное время текучести ПКС
Рис.

Эмульсии
Грубодисперсные системы типа Ж1/Ж2 (вода и масло)
1. Разбавленные, ϕ<1%
2. Концентрированные, ϕ=1-75%
Эмульсии
Грубодисперсные системы типа Ж1/Ж2 (вода и масло)
1. Разбавленные, ϕ<1%
2. Концентрированные, ϕ=1-75%

1 могут существовать без стабилизатора, во 2 и 3 обязательно наличие
1 могут существовать без стабилизатора, во 2 и 3 обязательно наличие
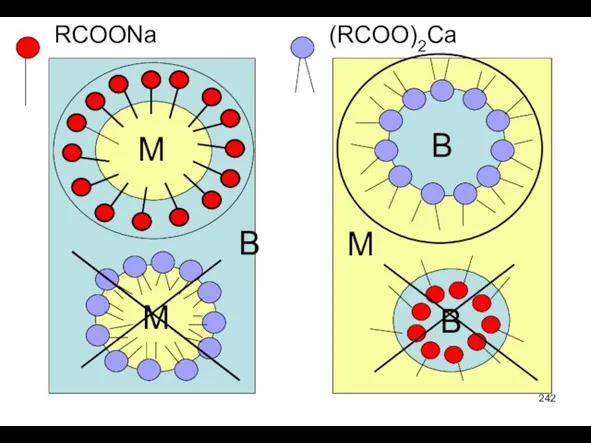
М
М
В
В
В
М
RCOONa
(RCOO)2Ca
М
М
В
В
В
М
RCOONa
(RCOO)2Ca
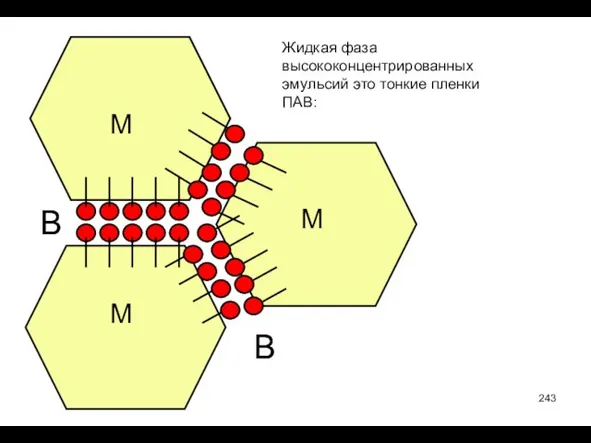
М
М
М
В
В
В
В
Жидкая фаза высококонцентрированных
эмульсий это тонкие пленки ПАВ:
М
М
М
В
В
В
В
Жидкая фаза высококонцентрированных
эмульсий это тонкие пленки ПАВ:

Получение эмульсий - диспергированием смеси жидкостей в присутствии ПАВ
Обращение фаз М/В↔В/М:
RCOONa
Получение эмульсий - диспергированием смеси жидкостей в присутствии ПАВ
Обращение фаз М/В↔В/М:
RCOONa

Системы типа Т1/Т2
Металлы и сплавы
Керамика
Наполненные полимеры (пластмассы, резина)
Минеральные ископаемые
Искусственные камни (бетон)
Органо-минеральные
Системы типа Т1/Т2
Металлы и сплавы
Керамика
Наполненные полимеры (пластмассы, резина)
Минеральные ископаемые
Искусственные камни (бетон)
Органо-минеральные

Системы типа Г/Ж и Г/Т
Аналог прямых эмульсий, газ вместо масла
Высококонцентрированные системы
Системы типа Г/Ж и Г/Т
Аналог прямых эмульсий, газ вместо масла
Высококонцентрированные системы

Аэрозоли системы типа Т/Г и Ж/Г
Т/Г – дым, пыль, Т/Ж –
Аэрозоли системы типа Т/Г и Ж/Г
Т/Г – дым, пыль, Т/Ж –

Размерные эффекты, нанотехнологии
Зависимость физических свойств от размера частиц и толщины пленок
Формула
Размерные эффекты, нанотехнологии
Зависимость физических свойств от размера частиц и толщины пленок
Формула

Полимеры
Полимеры
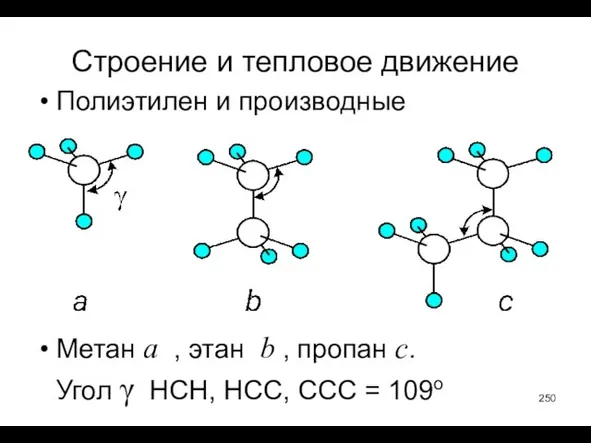
Строение и тепловое движение
Полиэтилен и производные
Метан a , этан b ,
Строение и тепловое движение
Полиэтилен и производные
Метан a , этан b ,
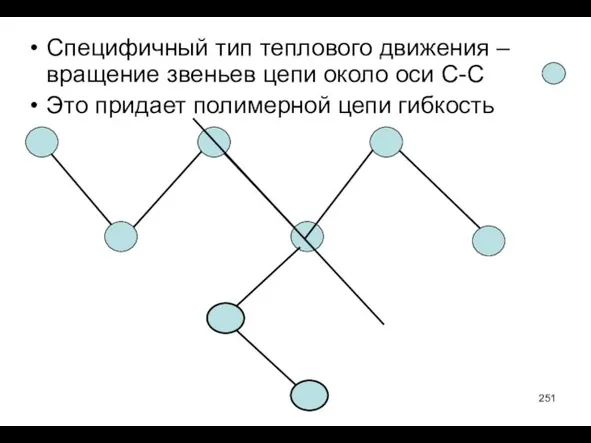
Специфичный тип теплового движения – вращение звеньев цепи около оси С-С
Это
Специфичный тип теплового движения – вращение звеньев цепи около оси С-С
Это
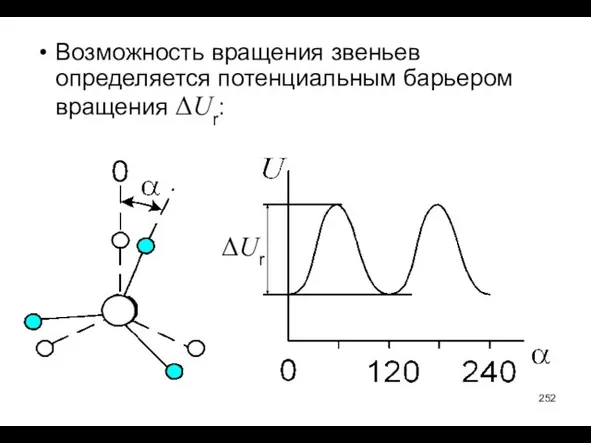
Возможность вращения звеньев определяется потенциальным барьером вращения ΔUr:
ΔUr
Возможность вращения звеньев определяется потенциальным барьером вращения ΔUr:
ΔUr
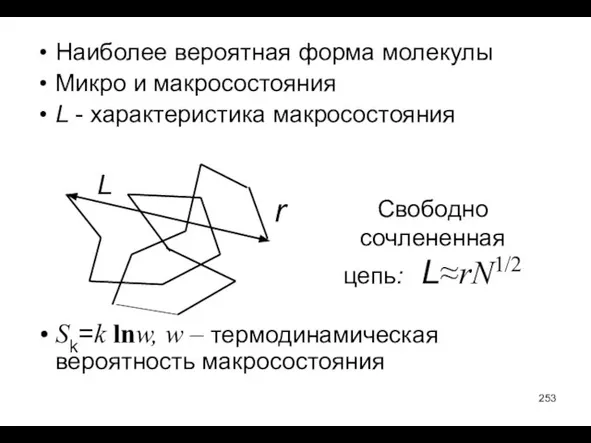
Наиболее вероятная форма молекулы
Микро и макросостояния
L - характеристика макросостояния
Sk=k lnw, w
Наиболее вероятная форма молекулы
Микро и макросостояния
L - характеристика макросостояния
Sk=k lnw, w

Свободная энергия F = U - TSk
Потенциальный барьер, вращательная подвижность звеньев
Свободная энергия F = U - TSk
Потенциальный барьер, вращательная подвижность звеньев
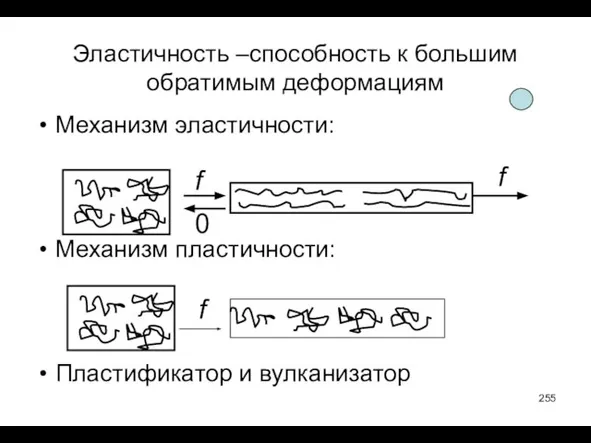
Эластичность –способность к большим обратимым деформациям
Механизм эластичности:
Механизм пластичности:
Пластификатор и вулканизатор
Эластичность –способность к большим обратимым деформациям
Механизм эластичности:
Механизм пластичности:
Пластификатор и вулканизатор
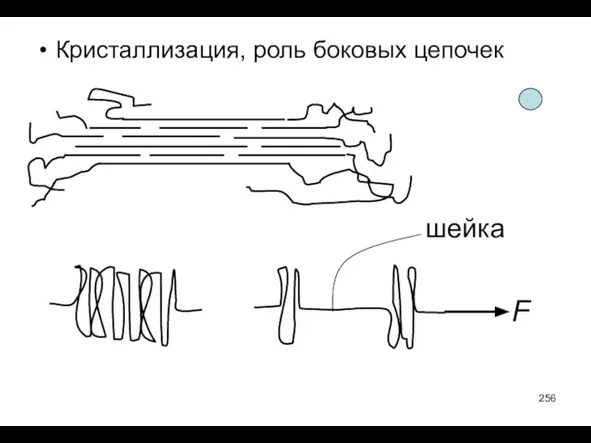
Кристаллизация, роль боковых цепочек
Кристаллизация, роль боковых цепочек
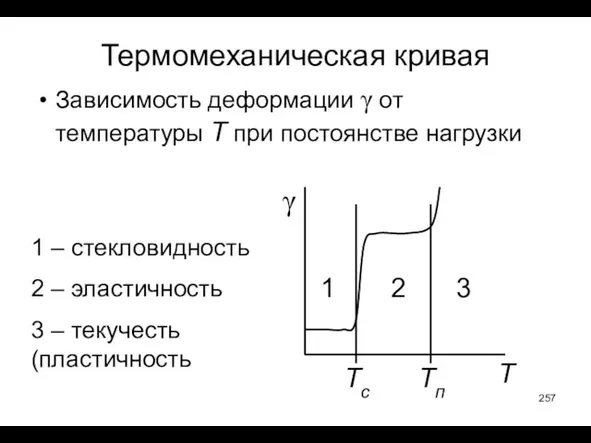
Термомеханическая кривая
Зависимость деформации γ от температуры T при постоянстве нагрузки
1 –
Термомеханическая кривая
Зависимость деформации γ от температуры T при постоянстве нагрузки
1 –

Растворы полимеров
Сходство и различие с коллоидами
Условие растворения
ΔG<0; ΔG =ΔH –TΔSk
Для полярных
Растворы полимеров
Сходство и различие с коллоидами
Условие растворения
ΔG<0; ΔG =ΔH –TΔSk
Для полярных
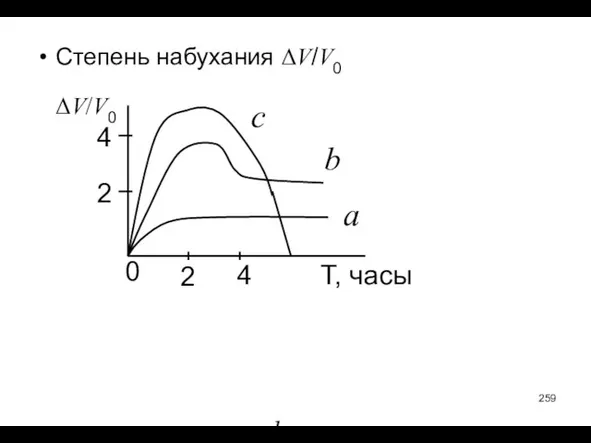
Степень набухания ΔV/V0
a – ограниченное, b - с частичным растворением, c
Степень набухания ΔV/V0
a – ограниченное, b - с частичным растворением, c

Вязкость растворов
В гидродинамическом отношении разбухшие клубки - сплошные частиц, применим вариант
Вязкость растворов
В гидродинамическом отношении разбухшие клубки - сплошные частиц, применим вариант

Слагаемое βϕ2 в формуле Эйнштейна – учет гидродинамического взаимодействия клубков. Это
Слагаемое βϕ2 в формуле Эйнштейна – учет гидродинамического взаимодействия клубков. Это

Полиэлектролиты
- полимеры, содержащие звенья, способные диссоциировать в растворе.
Поликислоты, полиоснования и амфотерные
Полиэлектролиты
- полимеры, содержащие звенья, способные диссоциировать в растворе.
Поликислоты, полиоснования и амфотерные
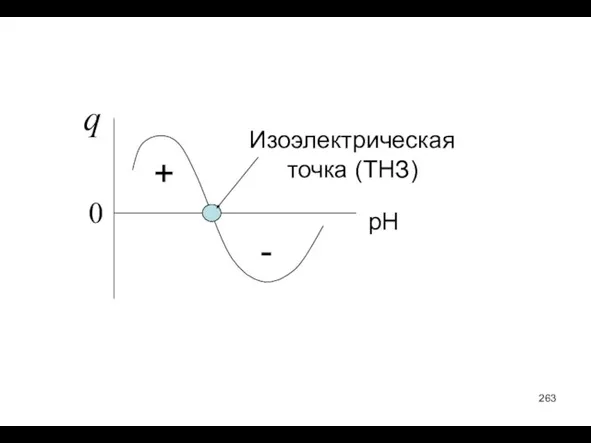
Изоэлектрическая точка (ТНЗ)
pH
q
0
+
-
Изоэлектрическая точка (ТНЗ)
pH
q
0
+
-

Вдали от ТНЗ отталкивание одноименно заряженных звеньев ведет к разбуханию клубка.
В
Вдали от ТНЗ отталкивание одноименно заряженных звеньев ведет к разбуханию клубка.
В
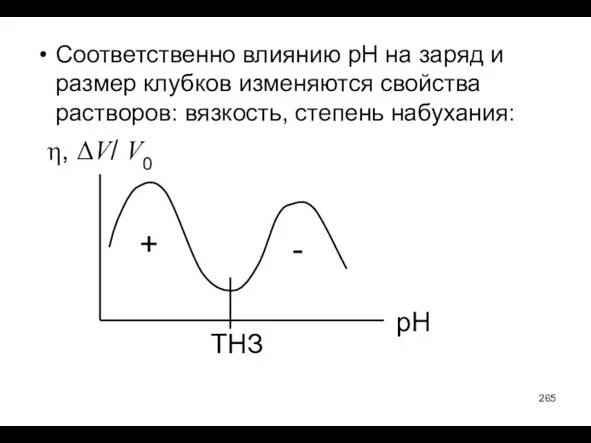
Соответственно влиянию рН на заряд и размер клубков изменяются свойства растворов:
Соответственно влиянию рН на заряд и размер клубков изменяются свойства растворов:
Похожие презентации
 Такой разный песок
Такой разный песок Әртүрлі заттардың кристаллдық тор энергиясын, сольваттану энергиясын, гидратация жылуын есептеу
Әртүрлі заттардың кристаллдық тор энергиясын, сольваттану энергиясын, гидратация жылуын есептеу Введение в общеобразовательную научную дисциплину "Химия"
Введение в общеобразовательную научную дисциплину "Химия" Коррозия металлов
Коррозия металлов
 Эволюция понятия “валентность”
Эволюция понятия “валентность” Металлы 4 группы А подгруппы
Металлы 4 группы А подгруппы Нефть. Состав нефти
Нефть. Состав нефти Мас-спектр кластерів вуглецю
Мас-спектр кластерів вуглецю Природные и попутные нефтяные газы
Природные и попутные нефтяные газы Ферроцен. Свойства, получение и применение
Ферроцен. Свойства, получение и применение Физические и химические явления
Физические и химические явления Жидкие кристаллы в технике
Жидкие кристаллы в технике Период становления химии
Период становления химии Презентация по Химии "Презентация Щелочные металлы" - скачать смотреть
Презентация по Химии "Презентация Щелочные металлы" - скачать смотреть  Термодинамика. 2 закон термодинамики. Энтропия
Термодинамика. 2 закон термодинамики. Энтропия Строительное материаловедение. (Лекции 1-2)
Строительное материаловедение. (Лекции 1-2) Есть ли сахар в банке с медом? или Определение качественного состава меда.
Есть ли сахар в банке с медом? или Определение качественного состава меда. Обобщение по теме неметаллы
Обобщение по теме неметаллы Carbon and the molecular diversity of life. (Chapter 4)
Carbon and the molecular diversity of life. (Chapter 4) Металлокомплексный катализ. (Лекция 16)
Металлокомплексный катализ. (Лекция 16) Биотехнология аминокислот и витаминов
Биотехнология аминокислот и витаминов Кислоты
Кислоты Презентация к уроку окружающего мира по теме: ,,Про воздух и про воду” Цель урока: знакомство учеников с богатствами природы - воздухом и водой, их свойствами. Задачи: Продолжить расширение и углубление представлений
Презентация к уроку окружающего мира по теме: ,,Про воздух и про воду” Цель урока: знакомство учеников с богатствами природы - воздухом и водой, их свойствами. Задачи: Продолжить расширение и углубление представлений  Коллоидные ПАВ
Коллоидные ПАВ Коррозия металлов
Коррозия металлов Сера
Сера Химическая термодинамика
Химическая термодинамика