- ПУТИ ОБМЕНА ОТДЕЛЬНЫХ АМИНОКИСЛОТ

Содержание
- 2. Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется ее бензольное кольцо, гликокетогенная. Тирозин
- 3. ФЕНИЛАЛАНИН ТИРОЗИН ОПК Белки Ацетоацетат Глюкоза Катехоламины Меланины Йодтиронины Дофамин Норадреналин Адреналин Нервная ткань Надпочечники Кожа
- 4. Тирозингидроксилаза (Fe2+) Адреналин Норадреналин Синтез катехоламинов (надпочечники, нервная ткань) О2 Н2О ДОФА-декарбоксилаза (В6) СО2 ДОФамингид- роксилаза
- 5. Значение катехоламинов ДОФамин – нейромедиатор среднего отдела мозга. Норадреналин – тормозный медиатор сим-патической нервной системы и
- 6. Болезнь Паркинсона – развивается при гипосек-реции дофамина в черной субстанции мозга (в среднем отделе мозга). Частота
- 7. Синтез меланинов (меланоциты) Кожа Волосы Радужная оболочка глаз CH CH2 NH2 CООH ОH CH CH2 NH2
- 8. Синтез йодтиронинов (щитовидная железа) Тиреоглобулин (ТГ) 2. Тирозин 2 J - 2е J20 (фиксация и окисление)
- 9. Синтез трийодтиронина (Т3) Монойодтирозин (3-йодтирозин) Дийодтирозин (3,5-дийодтирозин) Н2О ТГ 4. Трийодтиронин (Т3) 3 3 5 3
- 10. Синтез тетрайодтиронина (тироксина, Т4) Дийодтирозин (3,5-дийодтирозин) 3 5 Дийодтирозин (3,5-дийодтирозин) 3 5 Тетрайодитироксин (тироксин, Т4) 5.
- 11. Фенилаланин Тирозин n-гидроксифенилпируват Гомогентизиновая кислота Фумарилацетоацетат Фумарат Ацетоацетат Катаболизм фенилаланина и тирозина в печени О2 Н2О
- 12. Фен ПФ α - КГ Глу СН2 – С – СООН О Фенилпируват СН2 – СН
- 13. Белки (пищи и тканей) Фен Врожденные нарушения обмена ФЕН и ТИР Фенилпируват Фенилактат Фенилацетат Тир ДОФА
- 14. Фенилкетонурия – наследственное заболевание, наследуется по аутосомно-рецессивному типу, частота 1:10 тыс. новорожденных. дефект фермента фенилаланингидроксилазы. В
- 15. Проявления ФКУ – нарушения умственного и физического развития, судорожный синдром, нарушение пигментации. Больные не доживают до
- 16. Наследуется по аутосомно-рецессивному типу, частота 1:20 тыс. новорожденных. Причина метаболического нарушения - врожденный дефект тирозиназы, катализирующей
- 17. Наследуется по аутосомно-рецессивному типу, частота встречаемости – 2-5 : 1 млн. новорожденных. Причина заболевания - дефект
- 18. Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают 3 типа тирозинемии: 1) Тирозинемия
- 19. 2) Тирозинемия типа 2 (Синдром Рихнера –Ханхорта). Причиной является дефект фермента тирозинаминотранс-феразы. Для заболевания характерны поражения
- 20. Биологическая роль триптофана (незаменимая, гликокетогенная) Триптофан Серотонин (медиатор) Мелатонин (гормон) Белок (активные центры) Никотинамид (витамин РР)
- 21. Синтез серотонина (гладкая мускулатура, кишечник) и мелатонина (эпифиз) Триптофан NН СН2 СН СООН NН2 О2 Н2О
- 22. Обмен триптофана Триптофан Печень Никотинамид Серотонин Индолы Нервная система Бактерии желудочно- кишечного тракта Печень Глюкоза Кетоновые
- 23. Биологическая роль серотонина Стимулирует сокращения гладкой мускулатуры, перистальтику кишечника; Оказывает сосудосуживающее действием, регулирует АД, t, дыхание;
- 24. Синтез витамина РР Триптофан NН СН2 СН СООН NН2 О2 Н2О NН СН2 СН СООН NН2
- 25. Врожденное нарушение обмена триптофана - болезнь Хартнупа Возникает метаболический дефект связан с генетическим дефектом фермента триптофандиоксигеназы
- 26. NH2 N NH -CH2-CH-COOH NH2 NH3 N NH -CH=CH-COOH гистидаза Гистидин (гликопластическая, частично заменимая) Гистидин Уроканиновая
- 27. Биологическая роль гистамина Стимулирует секрецию желудочного сока, слюны; Повышает проницаемость капилляров, вызывает отеки; Снижает АД, но
- 28. Валин, лейцин, изолейцин Незаменимые аминокислоты Вал Лей Илей гликогенная (пропионил-КоА сукцинил-КоА глю) кетогенная гликокетогенная (ацетил-КоА +
- 29. Обмен аминокислот с разветвленной цепью Лейцин Изолейцин Валин α-Кетоизокапроат α-Кето-β-метилвалериат α-Кетоизовалериат Ацил-КоА- производ- ные жирных кислот
- 31. Скачать презентацию
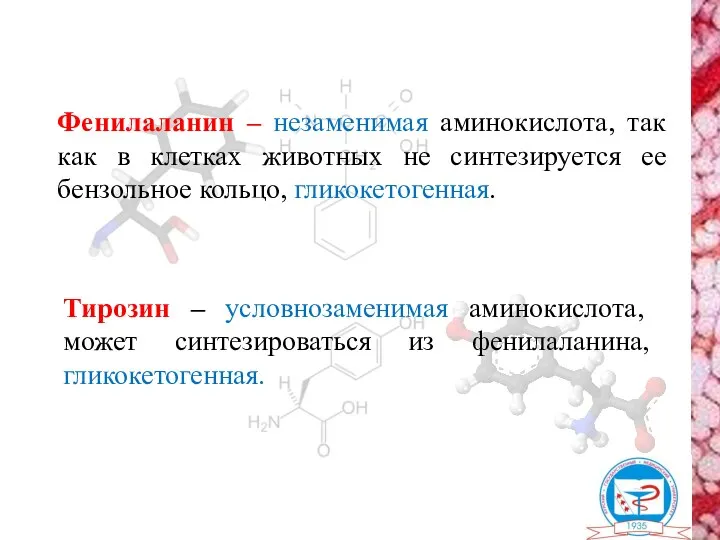
Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется
Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется
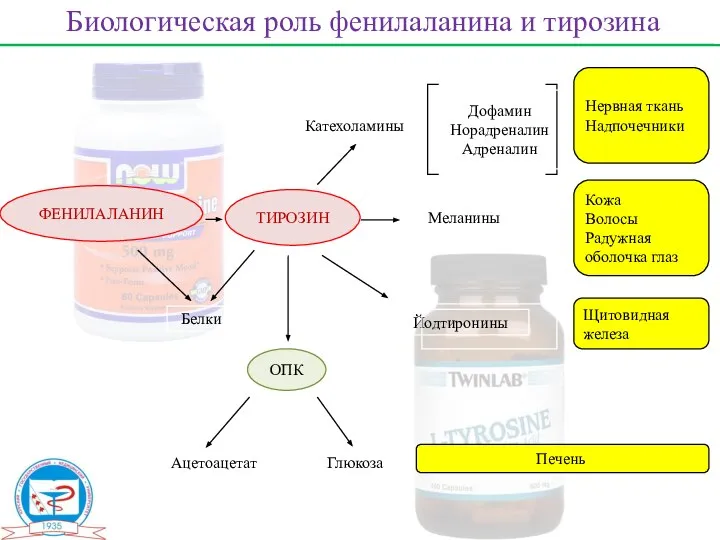
ФЕНИЛАЛАНИН
ТИРОЗИН
ОПК
Белки
Ацетоацетат
Глюкоза
Катехоламины
Меланины
Йодтиронины
Дофамин
Норадреналин
Адреналин
Нервная ткань
Надпочечники
Кожа
Волосы
Радужная оболочка глаз
Щитовидная железа
Печень
Биологическая роль фенилаланина и тирозина
ФЕНИЛАЛАНИН
ТИРОЗИН
ОПК
Белки
Ацетоацетат
Глюкоза
Катехоламины
Меланины
Йодтиронины
Дофамин
Норадреналин
Адреналин
Нервная ткань
Надпочечники
Кожа
Волосы
Радужная оболочка глаз
Щитовидная железа
Печень
Биологическая роль фенилаланина и тирозина
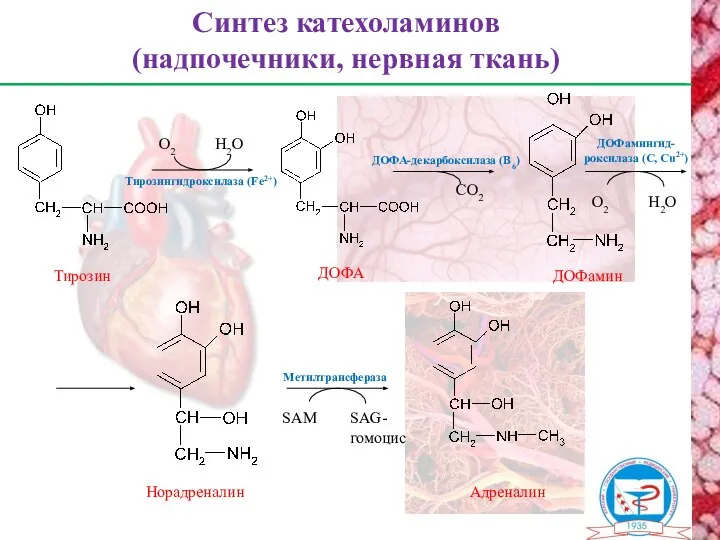
Тирозингидроксилаза (Fe2+)
Адреналин
Норадреналин
Синтез катехоламинов
(надпочечники, нервная ткань)
О2
Н2О
ДОФА-декарбоксилаза (В6)
СО2
ДОФамингид-
роксилаза (С, Сu2+)
О2
Н2О
Метилтрансфераза
SAM
SAG-гомоцис
Тирозингидроксилаза (Fe2+)
Адреналин
Норадреналин
Синтез катехоламинов
(надпочечники, нервная ткань)
О2
Н2О
ДОФА-декарбоксилаза (В6)
СО2
ДОФамингид-
роксилаза (С, Сu2+)
О2
Н2О
Метилтрансфераза
SAM
SAG-гомоцис

Значение катехоламинов
ДОФамин – нейромедиатор среднего отдела мозга.
Норадреналин – тормозный медиатор сим-патической
Значение катехоламинов
ДОФамин – нейромедиатор среднего отдела мозга.
Норадреналин – тормозный медиатор сим-патической

Болезнь Паркинсона – развивается при гипосек-реции дофамина в черной субстанции мозга
Болезнь Паркинсона – развивается при гипосек-реции дофамина в черной субстанции мозга
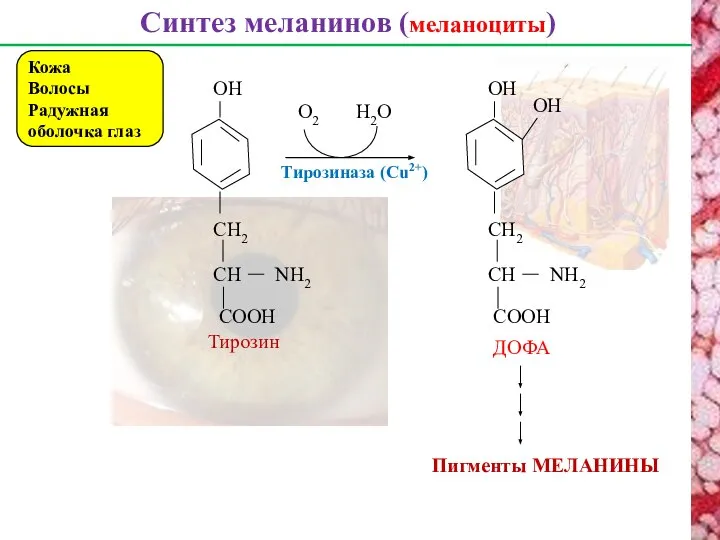
Синтез меланинов (меланоциты)
Кожа
Волосы
Радужная оболочка глаз
CH
CH2
NH2
CООH
ОH
CH
CH2
NH2
CООH
ОH
ОH
О2
ДОФА
Пигменты МЕЛАНИНЫ
Тирозин
Н2О
Тирозиназа (Cu2+)
Синтез меланинов (меланоциты)
Кожа
Волосы
Радужная оболочка глаз
CH
CH2
NH2
CООH
ОH
CH
CH2
NH2
CООH
ОH
ОH
О2
ДОФА
Пигменты МЕЛАНИНЫ
Тирозин
Н2О
Тирозиназа (Cu2+)
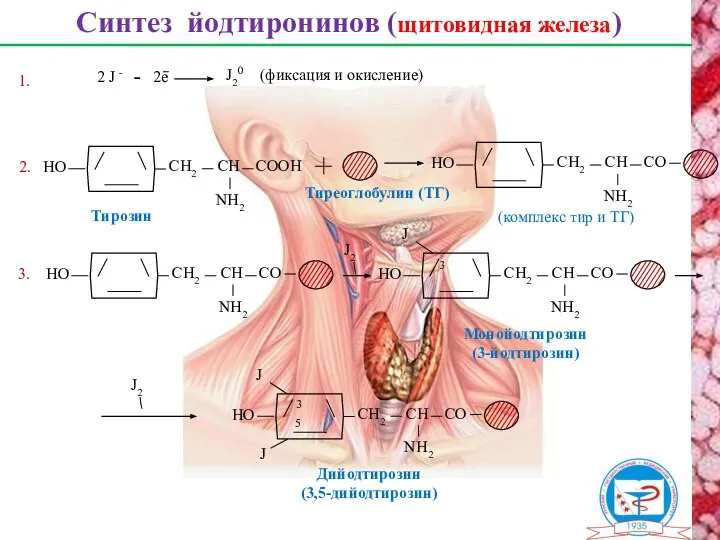
Синтез йодтиронинов (щитовидная железа)
Тиреоглобулин (ТГ)
2.
Тирозин
2 J -
2е
J20
(фиксация и окисление)
1.
3.
(комплекс тир
Синтез йодтиронинов (щитовидная железа)
Тиреоглобулин (ТГ)
2.
Тирозин
2 J -
2е
J20
(фиксация и окисление)
1.
3.
(комплекс тир
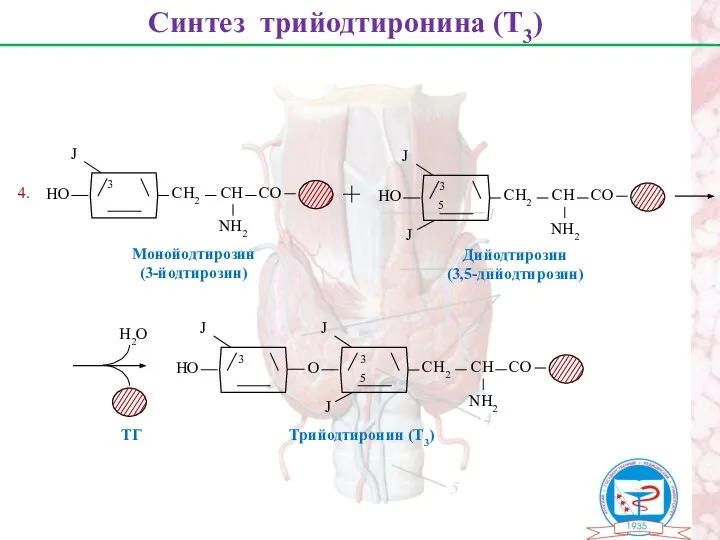
Синтез трийодтиронина (Т3)
Монойодтирозин
(3-йодтирозин)
Дийодтирозин
(3,5-дийодтирозин)
Н2О
ТГ
4.
Трийодтиронин (Т3)
3
3
5
3
5
3
Синтез трийодтиронина (Т3)
Монойодтирозин
(3-йодтирозин)
Дийодтирозин
(3,5-дийодтирозин)
Н2О
ТГ
4.
Трийодтиронин (Т3)
3
3
5
3
5
3
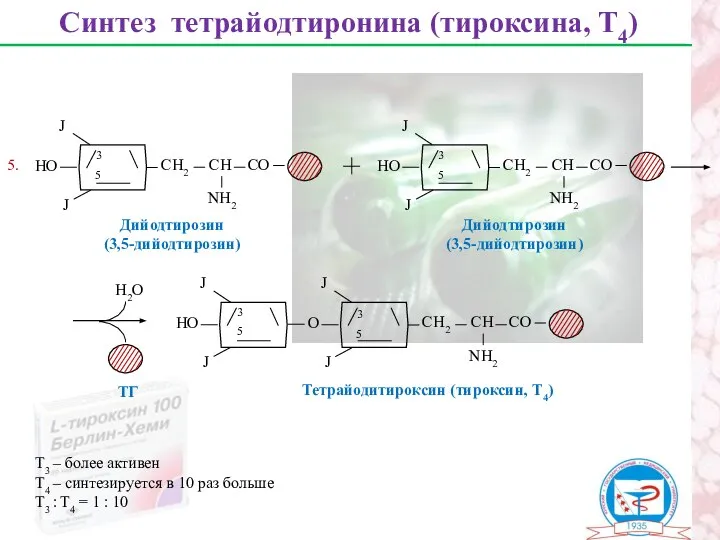
Синтез тетрайодтиронина (тироксина, Т4)
Дийодтирозин
(3,5-дийодтирозин)
3
5
Дийодтирозин
(3,5-дийодтирозин)
3
5
Тетрайодитироксин (тироксин, Т4)
5.
3
5
3
5
Т3 – более активен
Т4 – синтезируется
Синтез тетрайодтиронина (тироксина, Т4)
Дийодтирозин
(3,5-дийодтирозин)
3
5
Дийодтирозин
(3,5-дийодтирозин)
3
5
Тетрайодитироксин (тироксин, Т4)
5.
3
5
3
5
Т3 – более активен
Т4 – синтезируется
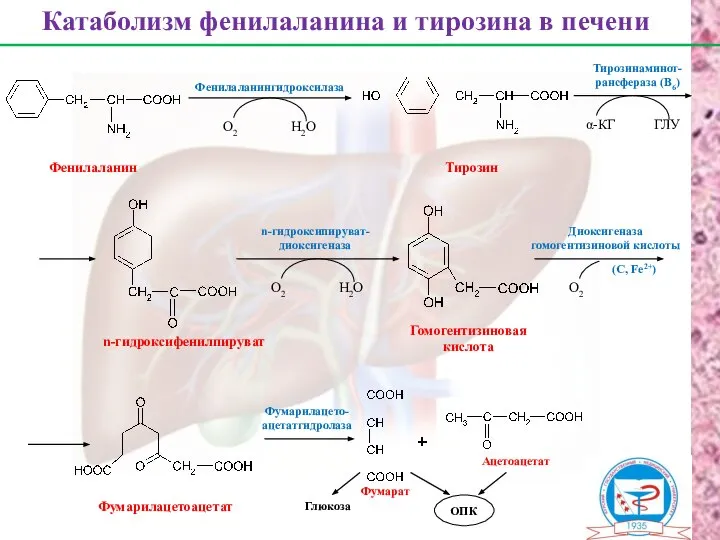
Фенилаланин
Тирозин
n-гидроксифенилпируват
Гомогентизиновая
кислота
Фумарилацетоацетат
Фумарат
Ацетоацетат
Катаболизм фенилаланина и тирозина в печени
О2
Н2О
Фенилаланингидроксилаза
α-КГ
ГЛУ
Тирозинаминот-
рансфераза (В6)
Фенилаланин
Тирозин
n-гидроксифенилпируват
Гомогентизиновая
кислота
Фумарилацетоацетат
Фумарат
Ацетоацетат
Катаболизм фенилаланина и тирозина в печени
О2
Н2О
Фенилаланингидроксилаза
α-КГ
ГЛУ
Тирозинаминот-
рансфераза (В6)
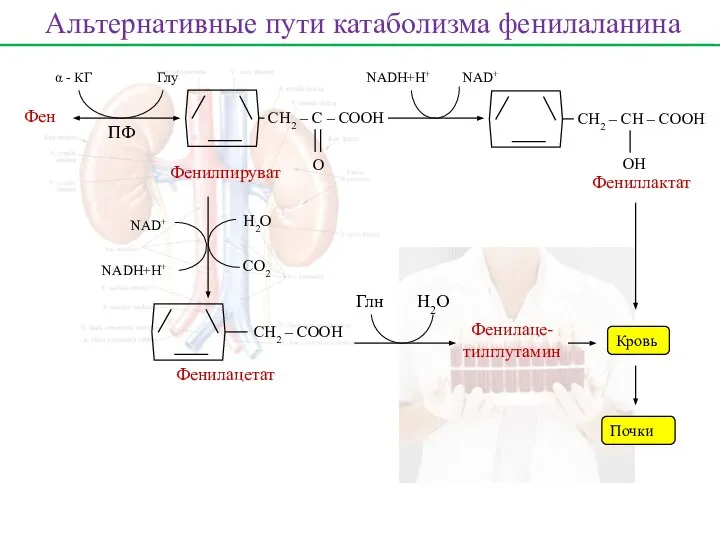
Фен
ПФ
α - КГ
Глу
СН2 – С – СООН
О
Фенилпируват
СН2 – СН – СООН
ОH
Фениллактат
Кровь
Почки
NADH+H+
NAD+
Фенилацетат
H2O
СО2
NADH+H+
NAD+
Фенилаце-
тилглутамин
H2O
Глн
Альтернативные
Фен
ПФ
α - КГ
Глу
СН2 – С – СООН
О
Фенилпируват
СН2 – СН – СООН
ОH
Фениллактат
Кровь
Почки
NADH+H+
NAD+
Фенилацетат
H2O
СО2
NADH+H+
NAD+
Фенилаце-
тилглутамин
H2O
Глн
Альтернативные
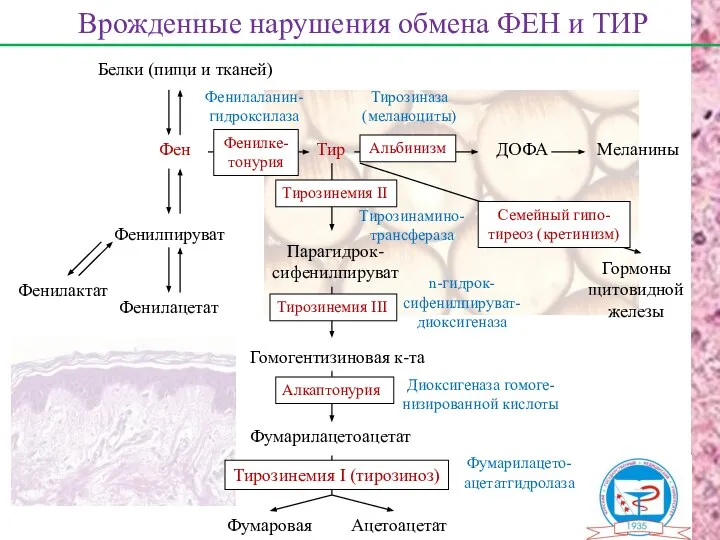
Белки (пищи и тканей)
Фен
Врожденные нарушения обмена ФЕН и ТИР
Фенилпируват
Фенилактат
Фенилацетат
Тир
ДОФА
Меланины
Гормоны
щитовидной
железы
Парагидрок-
сифенилпируват
n-гидрок-
сифенилпируват-
диоксигеназа
Фенилаланин-
гидроксилаза
Тирозиназа
(меланоциты)
Гомогентизиновая к-та
Диоксигеназа гомоге-
низированной
Белки (пищи и тканей)
Фен
Врожденные нарушения обмена ФЕН и ТИР
Фенилпируват
Фенилактат
Фенилацетат
Тир
ДОФА
Меланины
Гормоны
щитовидной
железы
Парагидрок-
сифенилпируват
n-гидрок-
сифенилпируват-
диоксигеназа
Фенилаланин-
гидроксилаза
Тирозиназа
(меланоциты)
Гомогентизиновая к-та
Диоксигеназа гомоге-
низированной

Фенилкетонурия – наследственное заболевание, наследуется по аутосомно-рецессивному типу, частота 1:10
Фенилкетонурия – наследственное заболевание, наследуется по аутосомно-рецессивному типу, частота 1:10

Проявления ФКУ – нарушения умственного и физического развития, судорожный синдром, нарушение
Проявления ФКУ – нарушения умственного и физического развития, судорожный синдром, нарушение

Наследуется по аутосомно-рецессивному типу,
частота 1:20 тыс. новорожденных.
Причина метаболического нарушения -
Наследуется по аутосомно-рецессивному типу,
частота 1:20 тыс. новорожденных.
Причина метаболического нарушения -

Наследуется по аутосомно-рецессивному типу, частота встречаемости – 2-5 : 1 млн.
Наследуется по аутосомно-рецессивному типу, частота встречаемости – 2-5 : 1 млн.
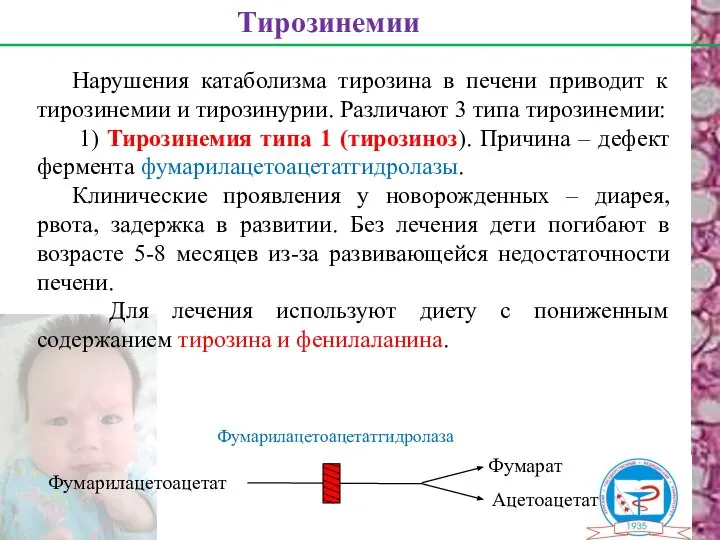
Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают
Нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Различают
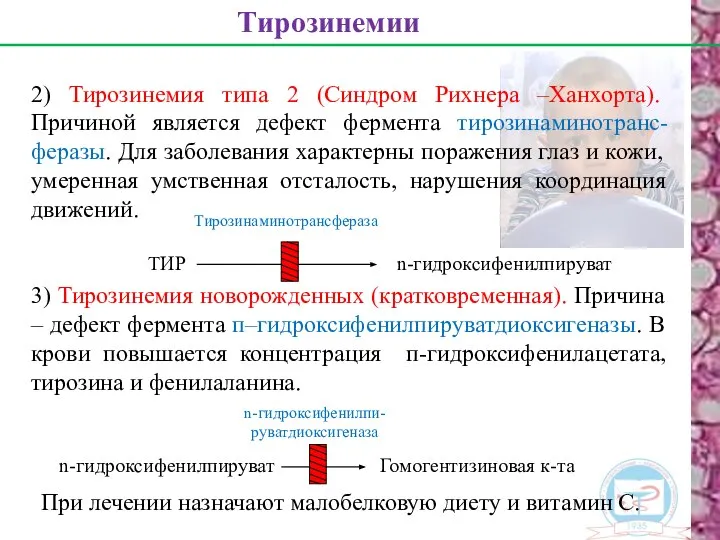
2) Тирозинемия типа 2 (Синдром Рихнера –Ханхорта). Причиной является дефект фермента
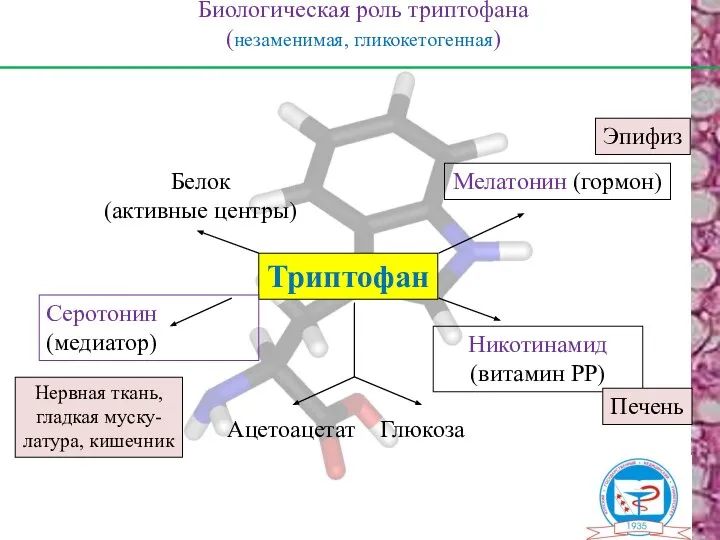
Биологическая роль триптофана
(незаменимая, гликокетогенная)
Триптофан
Серотонин
(медиатор)
Мелатонин (гормон)
Белок
(активные центры)
Никотинамид
(витамин РР)
Ацетоацетат
Глюкоза
Эпифиз
Печень
Нервная ткань,
гладкая муску-
латура,
Биологическая роль триптофана
(незаменимая, гликокетогенная)
Триптофан
Серотонин
(медиатор)
Мелатонин (гормон)
Белок
(активные центры)
Никотинамид
(витамин РР)
Ацетоацетат
Глюкоза
Эпифиз
Печень
Нервная ткань,
гладкая муску-
латура,
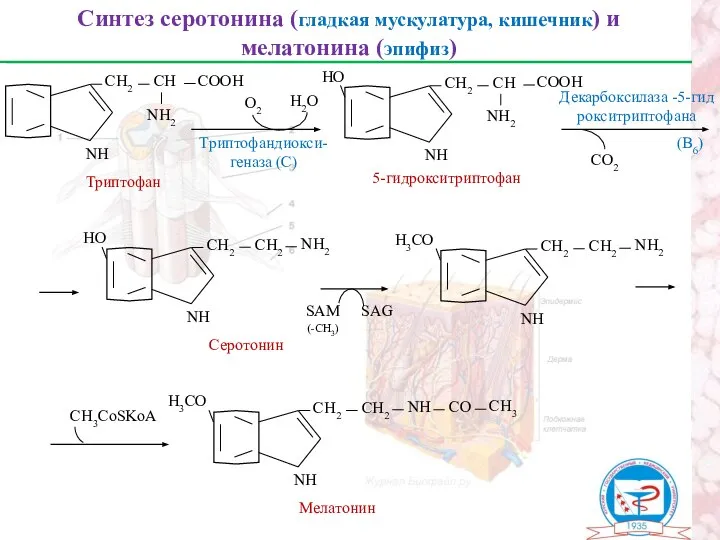
Синтез серотонина (гладкая мускулатура, кишечник) и мелатонина (эпифиз)
Триптофан
NН
СН2
СН
СООН
NН2
О2
Н2О
NН
СН2
СН
СООН
NН2
НО
5-гидрокситриптофан
СО2
Декарбоксилаза -5-гид
рокситриптофана
NН
СН2
СН2
NН2
НО
SAM
(-CH3)
NН
СН2
СН2
NН2
Н3СО
Серотонин
СН3CoSKoA
NН
СН2
СН2
NН
Н3СО
СO
СН3
Мелатонин
Триптофандиокси-
геназа (С)
SAG
(В6)
Синтез серотонина (гладкая мускулатура, кишечник) и мелатонина (эпифиз)
Триптофан
NН
СН2
СН
СООН
NН2
О2
Н2О
NН
СН2
СН
СООН
NН2
НО
5-гидрокситриптофан
СО2
Декарбоксилаза -5-гид
рокситриптофана
NН
СН2
СН2
NН2
НО
SAM
(-CH3)
NН
СН2
СН2
NН2
Н3СО
Серотонин
СН3CoSKoA
NН
СН2
СН2
NН
Н3СО
СO
СН3
Мелатонин
Триптофандиокси-
геназа (С)
SAG
(В6)
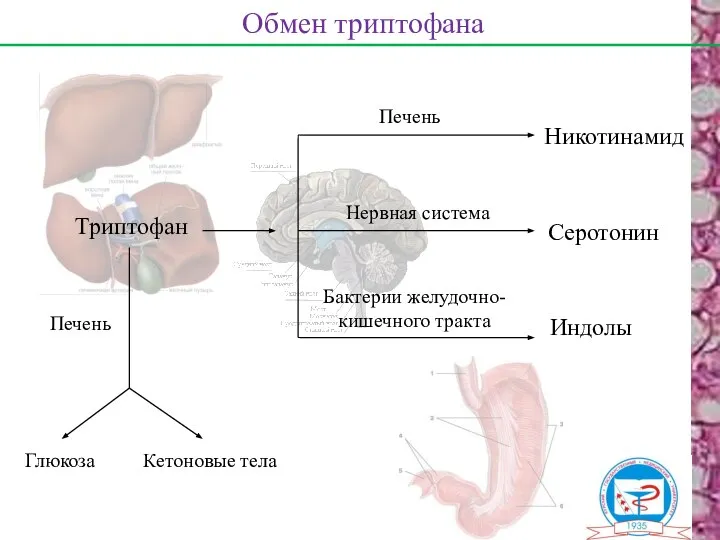
Обмен триптофана
Триптофан
Печень
Никотинамид
Серотонин
Индолы
Нервная система
Бактерии желудочно-
кишечного тракта
Печень
Глюкоза
Кетоновые тела
Обмен триптофана
Триптофан
Печень
Никотинамид
Серотонин
Индолы
Нервная система
Бактерии желудочно-
кишечного тракта
Печень
Глюкоза
Кетоновые тела
Биологическая роль серотонина
Стимулирует сокращения гладкой мускулатуры, перистальтику кишечника;
Оказывает сосудосуживающее действием, регулирует
Биологическая роль серотонина
Стимулирует сокращения гладкой мускулатуры, перистальтику кишечника;
Оказывает сосудосуживающее действием, регулирует
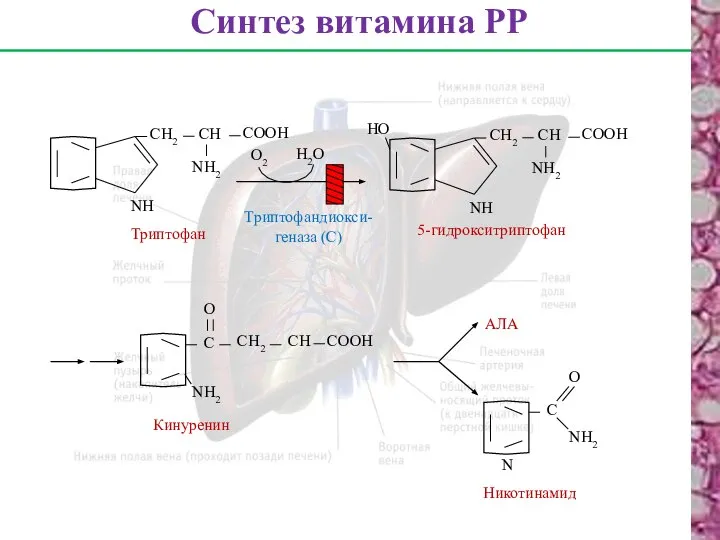
Синтез витамина РР
Триптофан
NН
СН2
СН
СООН
NН2
О2
Н2О
NН
СН2
СН
СООН
NН2
НО
5-гидрокситриптофан
Триптофандиокси-
геназа (С)
С
СН2
СН
СООН
О
NН2
Кинуренин
АЛА
С
О
N
NН2
Никотинамид
Синтез витамина РР
Триптофан
NН
СН2
СН
СООН
NН2
О2
Н2О
NН
СН2
СН
СООН
NН2
НО
5-гидрокситриптофан
Триптофандиокси-
геназа (С)
С
СН2
СН
СООН
О
NН2
Кинуренин
АЛА
С
О
N
NН2
Никотинамид

Врожденное нарушение обмена триптофана -
болезнь Хартнупа
Возникает метаболический дефект связан с
Врожденное нарушение обмена триптофана -
болезнь Хартнупа
Возникает метаболический дефект связан с
NH2
N
NH
-CH2-CH-COOH
NH2
NH3
N
NH
-CH=CH-COOH
гистидаза
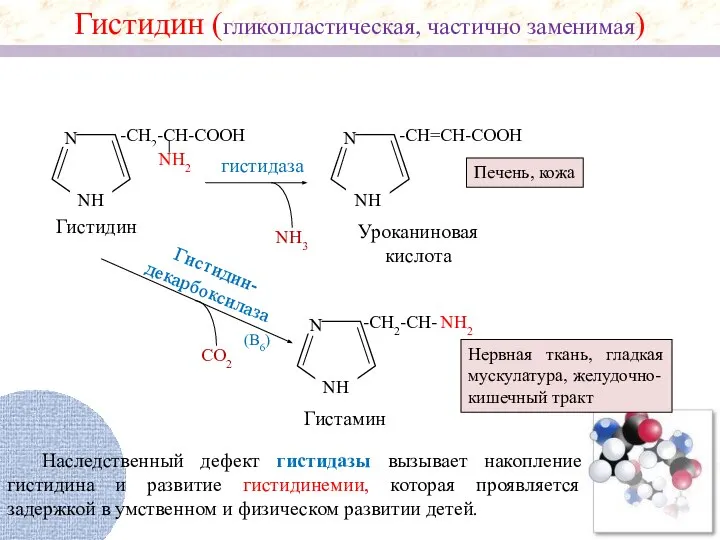
Гистидин (гликопластическая, частично заменимая)
Гистидин
Уроканиновая кислота
Печень, кожа
Гистидин-
декарбоксилаза
СО2
N
NH
-CH2-CH-
(В6)
Гистамин
Нервная ткань, гладкая мускулатура, желудочно-кишечный тракт
Наследственный
NH2
N
NH
-CH2-CH-COOH
NH2
NH3
N
NH
-CH=CH-COOH
гистидаза
Гистидин (гликопластическая, частично заменимая)
Гистидин
Уроканиновая кислота
Печень, кожа
Гистидин-
декарбоксилаза
СО2
N
NH
-CH2-CH-
(В6)
Гистамин
Нервная ткань, гладкая мускулатура, желудочно-кишечный тракт
Наследственный

Биологическая роль гистамина
Стимулирует секрецию желудочного сока, слюны;
Повышает проницаемость капилляров, вызывает отеки;
Снижает
Биологическая роль гистамина
Стимулирует секрецию желудочного сока, слюны;
Повышает проницаемость капилляров, вызывает отеки;
Снижает
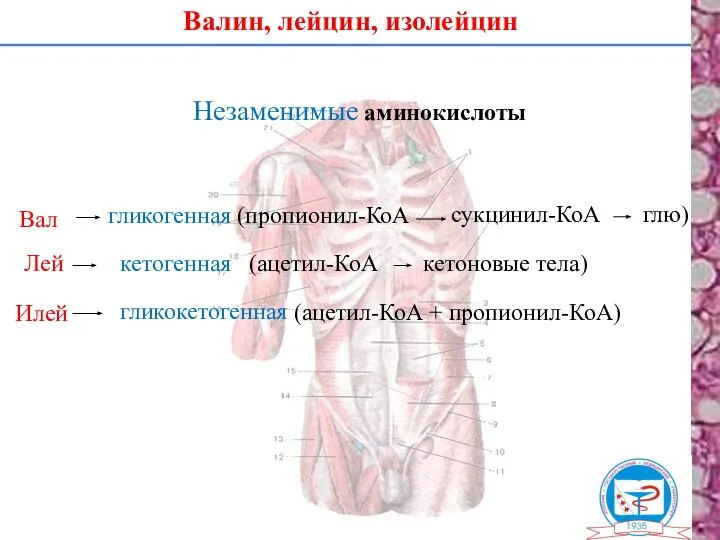
Валин, лейцин, изолейцин
Незаменимые аминокислоты
Вал
Лей
Илей
гликогенная (пропионил-КоА
сукцинил-КоА
глю)
кетогенная
гликокетогенная
(ацетил-КоА + пропионил-КоА)
(ацетил-КоА
кетоновые тела)
Валин, лейцин, изолейцин
Незаменимые аминокислоты
Вал
Лей
Илей
гликогенная (пропионил-КоА
сукцинил-КоА
глю)
кетогенная
гликокетогенная
(ацетил-КоА + пропионил-КоА)
(ацетил-КоА
кетоновые тела)
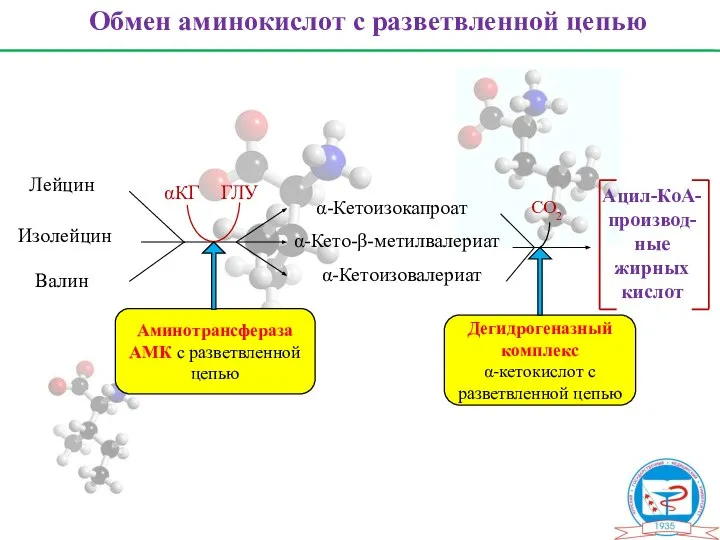
Обмен аминокислот с разветвленной цепью
Лейцин
Изолейцин
Валин
α-Кетоизокапроат
α-Кето-β-метилвалериат
α-Кетоизовалериат
Ацил-КоА-
производ-
ные
жирных
кислот
CО2
αКГ
ГЛУ
Аминотрансфераза
АМК с разветвленной цепью
Дегидрогеназный комплекс
α-кетокислот с разветвленной
Обмен аминокислот с разветвленной цепью
Лейцин
Изолейцин
Валин
α-Кетоизокапроат
α-Кето-β-метилвалериат
α-Кетоизовалериат
Ацил-КоА-
производ-
ные
жирных
кислот
CО2
αКГ
ГЛУ
Аминотрансфераза
АМК с разветвленной цепью
Дегидрогеназный комплекс
α-кетокислот с разветвленной
 Алкілування ізобутану бутенами
Алкілування ізобутану бутенами Роль минеральных веществ
Роль минеральных веществ Галогены VII группы. Биологическая роль и применение в медицине
Галогены VII группы. Биологическая роль и применение в медицине Нетканые материалы из химических волокон Выполнила Гехт Ольга Алексеевна учитель технологии МАОУ СОШ №88 г. Тюмень
Нетканые материалы из химических волокон Выполнила Гехт Ольга Алексеевна учитель технологии МАОУ СОШ №88 г. Тюмень Почвоведение. Введение
Почвоведение. Введение Кометика Liv Delano - презентация_
Кометика Liv Delano - презентация_ Калитина Тамара Михайловна учитель экологии, биологии МОУ СОШ №3 и учитель химии МОУ СОШ №2 с.Александров-Гай Саратовской области
Калитина Тамара Михайловна учитель экологии, биологии МОУ СОШ №3 и учитель химии МОУ СОШ №2 с.Александров-Гай Саратовской области  Химия муравьиной кислоты
Химия муравьиной кислоты La notion de catabolisme et d’anabolisme. La bioenergetique. La chaine mitochondriale de transfert d’electrons
La notion de catabolisme et d’anabolisme. La bioenergetique. La chaine mitochondriale de transfert d’electrons Изотопный обмен
Изотопный обмен 8 шагов к кристаллу
8 шагов к кристаллу Углеволокно (карбон)
Углеволокно (карбон) Презентация по Химии "Номенклатура алканов разветвлённого строения" - скачать смотреть
Презентация по Химии "Номенклатура алканов разветвлённого строения" - скачать смотреть  Оксиди. Кислоти
Оксиди. Кислоти Зависимость свойств веществ от их строения. Химическая связь. Основные виды химической связи
Зависимость свойств веществ от их строения. Химическая связь. Основные виды химической связи Магний и его соединения
Магний и его соединения 9 класс Урок №9. Углеводы. Составитель презентации – учитель химии МОУ СОШ г. Холма Насонова Т.А.
9 класс Урок №9. Углеводы. Составитель презентации – учитель химии МОУ СОШ г. Холма Насонова Т.А.  Строение атома
Строение атома Неметаллические материалы
Неметаллические материалы Круговорот азота в природе. N2
Круговорот азота в природе. N2 Алканы. Определение. Общая формула класса углеводородов
Алканы. Определение. Общая формула класса углеводородов Сульфатная кислота. Сульфаты
Сульфатная кислота. Сульфаты Морозостойкая эпоксидная клеевая композиция марки УП-10-04М
Морозостойкая эпоксидная клеевая композиция марки УП-10-04М Искусственные материалы. Пластмассы
Искусственные материалы. Пластмассы Термодинамика. Законы термодинамики
Термодинамика. Законы термодинамики Феноли. Застосування в медицині та повсякденному житті
Феноли. Застосування в медицині та повсякденному житті Вещества и их системы. Смешивание растворов
Вещества и их системы. Смешивание растворов Сложные эфиры. Жиры. Мыла
Сложные эфиры. Жиры. Мыла