- Теория нитрования

Содержание
- 2. Лекция 1
- 3. Принципиальная схема цеха по получению нитросоединений Прием и хранение сырья Подготовка компонентов Реакционный узел Очистка продукта
- 4. РЕАКЦИОННЫЙ УЗЕЛ (СТАДИЯ НИТРОВАНИЯ) Периодические и непрерывные процессы. Достоинства и недостатки. Зависимость от производительности системы. Реакторы
- 5. Тип реакции нитрования Электрофильное замещение Радикальный механизм Ион-радикальный механизм Нуклеофильное замещение Щелочное нитрование Косвенные методы введения
- 6. Нитрующие системы и нитрующие агенты По кислотности и «активности» (кислотные и мало кислые либо нейтральные) По
- 7. Уксусно-ангидридные нитрующие смеси
- 8. ТНПДМ ТНГУ Нитрующая активность уксусно-ангидридных смесей на примере циклических мочевин
- 9. Нитрующие смеси на кислотной основе
- 10. Механизм нитрования Механизм реакции – совокупность элементарных процессов Методы изучения: изучение кинетики процессов, физико-химическине (спектральные) и
- 11. В настоящее время общепринятым является механизм электофильного нитрования с участием иона нитрония NO2+ (А.И. Титов, К.
- 12. Образование иона нитрония В азотной кислоте В безводной кислоте 2HNO3 ↔ H2NO3+ + NO3- ↔ ↔
- 13. Образование нитроний иона в серно-азотных смесях Доказательства: Кинетические Спектральные (ЯМР, СКР – 1400 см-1) Электро-химические –
- 14. Лекция 2
- 15. Зависимость т.. пл. азотной кислоты от концентрации А - -42,3°С, 33%; Б - -42°С, 70%; С
- 16. Серная кислота и ее гидраты
- 17. Азотная кислота и ее гидраты
- 18. Ионно-молекулярный состав серной кислоты H2SO4 + 2H2O ↔ H3O+ * H2O * HSO4- H3O+ * H2O
- 19. Функции кислотности Теория Бренстеда – Лоури. Согласно теории Бренстеда – Лоури кислота рассматривается как вещество, поставляющее
- 20. Физический смысл и меры основности в газовой фазе Основностью в газовой фазе называют свободную энергию (
- 21. Протонирование слабых органических оснований в водных растворах кислот. Протонирование многих слабых органических оснований происходит в достаточно
- 22. В зависимости от концентрации минеральной кислоты, характер специфической сольватации непрерывно меняется. Избыточные протоны, имеющиеся в водных
- 23. Гаммет и Дейруп: серия органических соединений, обладающие максимально сходной молекулярной структурой, так называемых кислотно-основных индикаторов. Протонирование
- 25. Функция кислотности Н0 с успехом применяется для количественного сравнения слабых оснований, протонирование которых может быть выражено
- 26. 100% H2SO4 97% H2SO4
- 27. Кроме функций Нo и Hr существуют функции построенные на других рядах индикаторов: амидах (Ha), индолах и
- 28. HNO3+ 2 Н2SO4 ↔ H3O+ + NO2+ + 2HSO4- где Hr и Mc функции кислотности Log(I)=[NO2+]/[HNO3]
- 30. Лекция 3
- 32. Химическая кинетика и химическая динамика: иерархия времён
- 33. Смесь №1 54,53% HNO3, 20,31% H2SO4, 25,16% H2O Смесь №2 3,6% HNO3, 67,47% H2SO4, 28,93% H2O
- 35. Пример кинетической кривой нитрования в серно-азотной нитрующей смеси, содержащей 73,5% серную кислоту, при 60оС.
- 36. +NO2+ k1 +H+ k-1 +NO2+ k2 +H+ k-2
- 37. 7.6.Схема нитрования биурета +NO2+ k1 +H+ k-1 +NO2+ k2 +H+ k-2 - степень ионизации HNO3 Lg
- 38. Примеры обработки кинетических данных по уравнению для последовательных реакций 80 % 85 % 93 % 20
- 41. Определение pKa 4-фенил-1,2,4-триазол-5-она
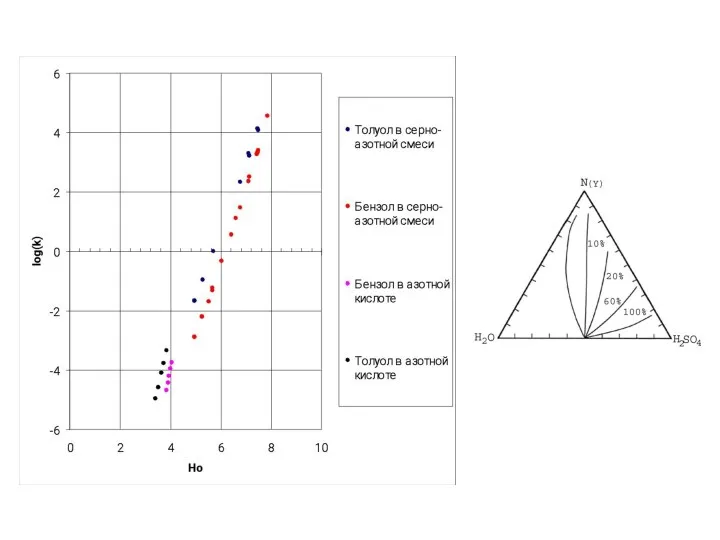
- 42. Зависимость констант скоростей нитрования ТО от (Hr + lgaH2O) в 65 – 80% среде серной кислоты.
- 44. Рис. Зависимость эффективной константы скорости к2эф при нитровании 2,4-динитротолуола от концентраций HNO3 и H2SO4
- 46. ТЕПЛОВЫЕ ЭФФЕКТЫ ПРИ НИТРОВАНИИ Тепловой баланс. Q приход = Q1 + Q2 + Q3 Приход Q1
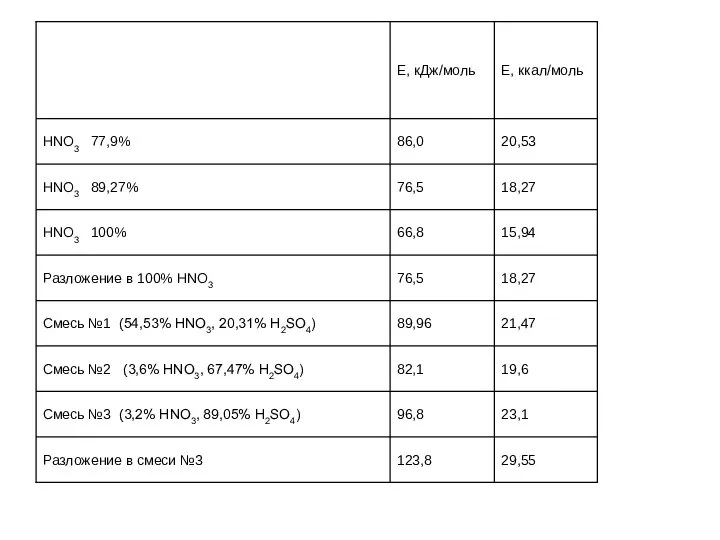
- 47. Суммарный тепловой эффект нитрования при получении мононитротолуола ~ 123 кДж/мол (29 ккал/моль) динитротолуола ~ 139 кДж/моль
- 48. Тепловой эффект основных реакций идущих при получении тринитротолуола можно рассчитать по формулам Q = a +
- 49. rD = β∑* (C2/γ –C1), где β∑ -общее сопротивление диффузии Если скорость химической реакции значительно ниже,
- 50. Кинетика гетерогенных процессов Процессы диффузии С2 С2.0 С1.0 С1 D –коэффициент диффузии rD =D *dc/dx мол/м2*с
- 51. Влияние азотистой кислоты В водных растворах H2SO4 и HClO4 (с концентрацией менее 50%) HNO2 присутствует преимущественно
- 52. Изучение кинетики каталитического процесса строго доказало первоначальное нитрозирование с последующим быстрым окислением С-нитрозосоединения и регенерацией HNO2.
- 53. Последующие исследования показали, что при каталитическом действии низших оксидов азота не всегда происходит нитрозирование. Этот явление
- 54. В 70-80-е годы образование относительно стабильных катион-радикалов (КР) спектральными методами или в форме солей было обнаружено
- 56. Скачать презентацию

Лекция 1
Лекция 1
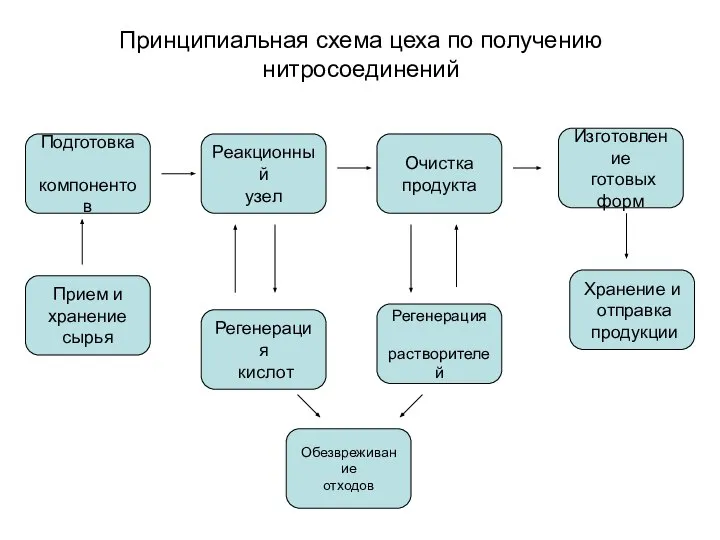
Принципиальная схема цеха по получению нитросоединений
Прием и
хранение
сырья
Подготовка
компонентов
Реакционный
узел
Очистка
Принципиальная схема цеха по получению нитросоединений
Прием и
хранение
сырья
Подготовка
компонентов
Реакционный
узел
Очистка
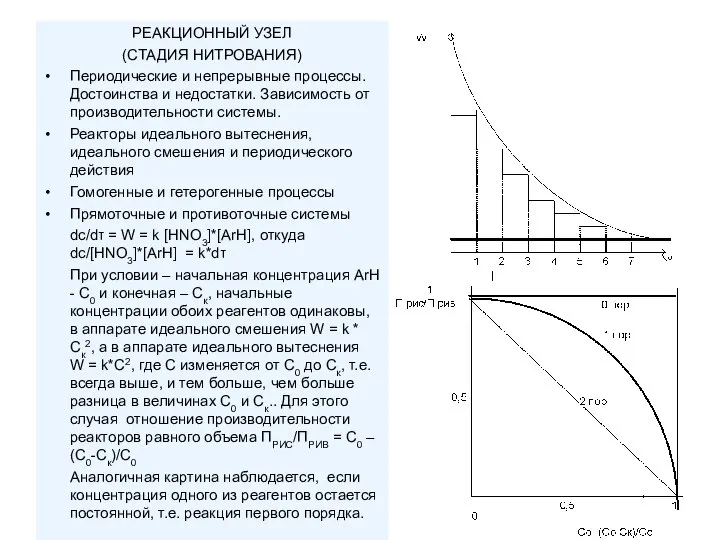
РЕАКЦИОННЫЙ УЗЕЛ
(СТАДИЯ НИТРОВАНИЯ)
Периодические и непрерывные процессы. Достоинства и недостатки. Зависимость
РЕАКЦИОННЫЙ УЗЕЛ
(СТАДИЯ НИТРОВАНИЯ)
Периодические и непрерывные процессы. Достоинства и недостатки. Зависимость
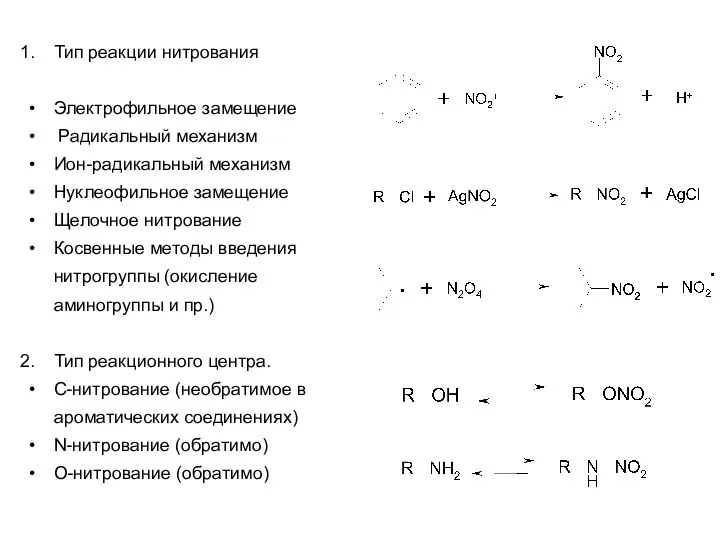
Тип реакции нитрования
Электрофильное замещение
Радикальный механизм
Ион-радикальный механизм
Нуклеофильное замещение
Щелочное нитрование
Косвенные методы введения
Тип реакции нитрования
Электрофильное замещение
Радикальный механизм
Ион-радикальный механизм
Нуклеофильное замещение
Щелочное нитрование
Косвенные методы введения
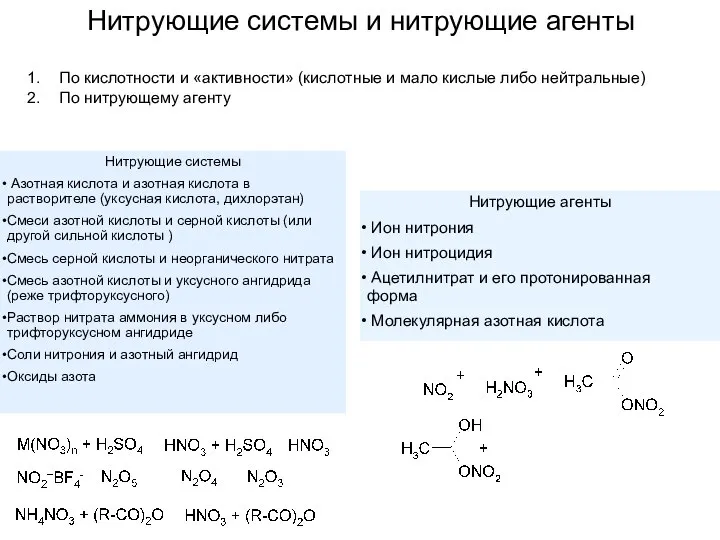
Нитрующие системы и нитрующие агенты
По кислотности и «активности» (кислотные и мало
Нитрующие системы и нитрующие агенты
По кислотности и «активности» (кислотные и мало
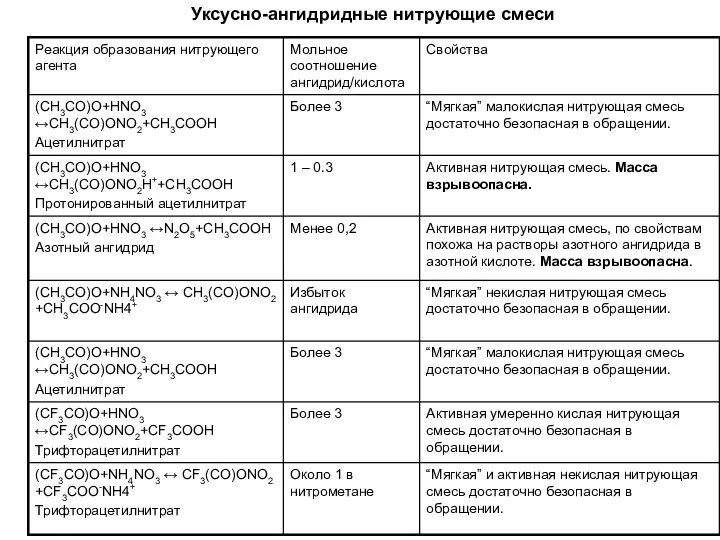
Уксусно-ангидридные нитрующие смеси
Уксусно-ангидридные нитрующие смеси
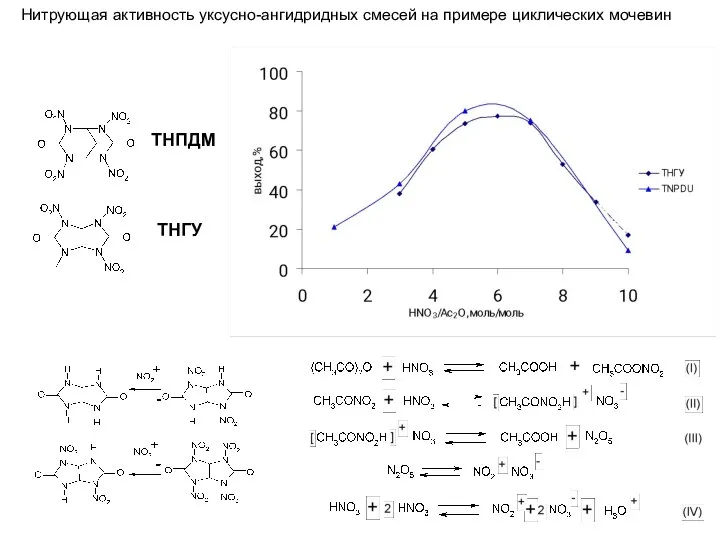
ТНПДМ
ТНГУ
Нитрующая активность уксусно-ангидридных смесей на примере циклических мочевин
ТНПДМ
ТНГУ
Нитрующая активность уксусно-ангидридных смесей на примере циклических мочевин

Нитрующие смеси на кислотной основе
Нитрующие смеси на кислотной основе

Механизм нитрования
Механизм реакции – совокупность элементарных процессов
Методы изучения: изучение кинетики процессов,
Механизм нитрования
Механизм реакции – совокупность элементарных процессов
Методы изучения: изучение кинетики процессов,

В настоящее время общепринятым является механизм электофильного нитрования с участием иона
В настоящее время общепринятым является механизм электофильного нитрования с участием иона
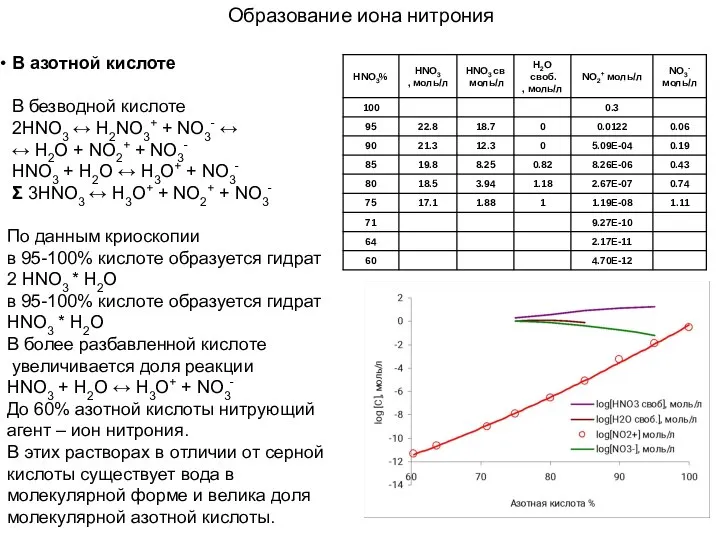
Образование иона нитрония
В азотной кислоте
В безводной кислоте
2HNO3
Образование иона нитрония
В азотной кислоте
В безводной кислоте
2HNO3
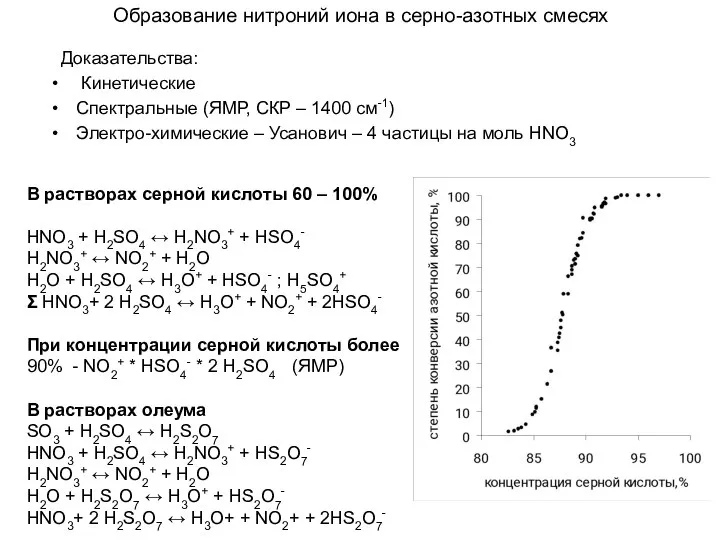
Образование нитроний иона в серно-азотных смесях
Доказательства:
Кинетические
Спектральные (ЯМР, СКР –
Образование нитроний иона в серно-азотных смесях
Доказательства:
Кинетические
Спектральные (ЯМР, СКР –

Лекция 2
Лекция 2
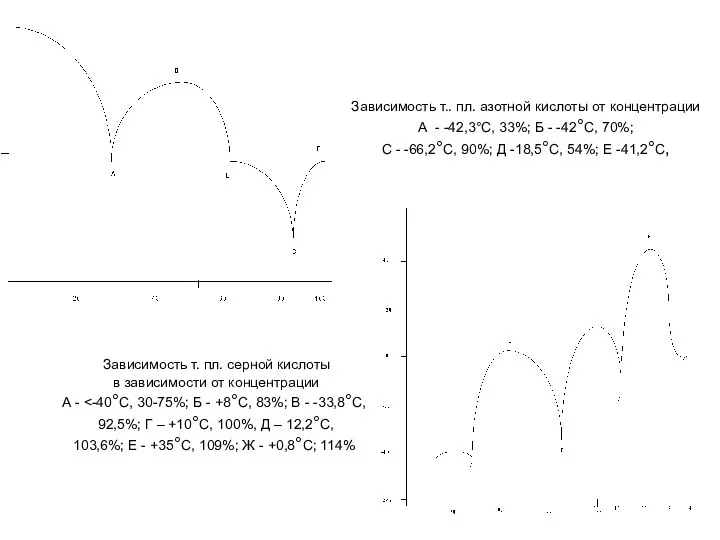
Зависимость т.. пл. азотной кислоты от концентрации
А - -42,3°С, 33%; Б
Зависимость т.. пл. азотной кислоты от концентрации
А - -42,3°С, 33%; Б
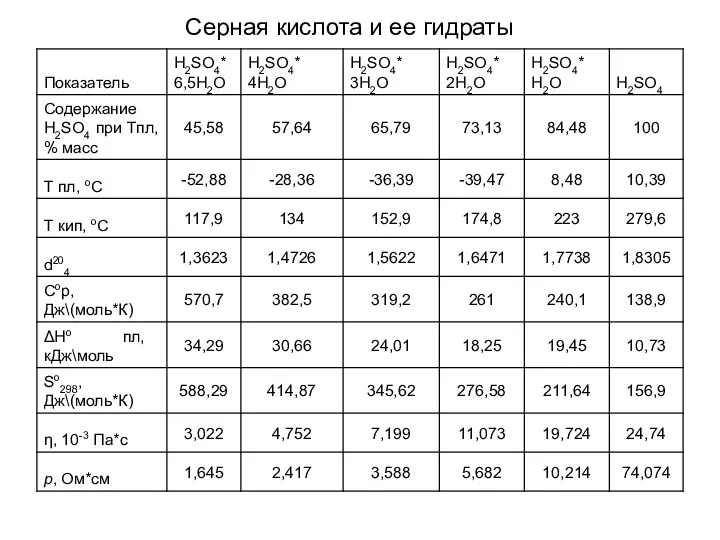
Серная кислота и ее гидраты
Серная кислота и ее гидраты
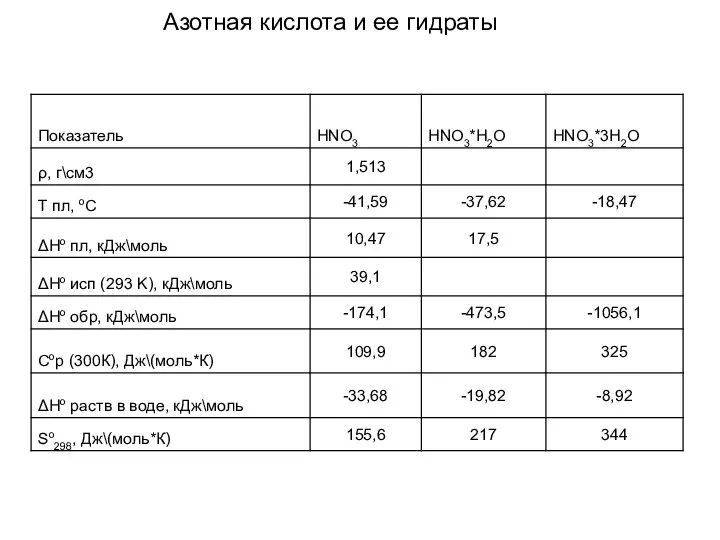
Азотная кислота и ее гидраты
Азотная кислота и ее гидраты
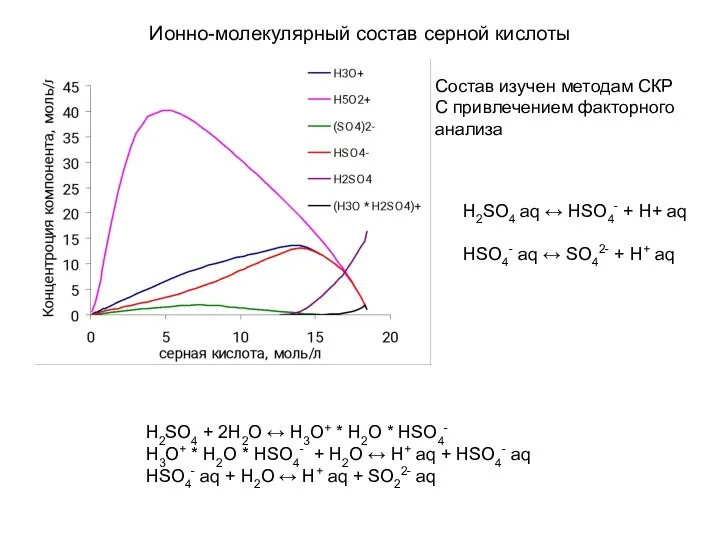
Ионно-молекулярный состав серной кислоты
H2SO4 + 2H2O ↔ H3O+ * H2O
Ионно-молекулярный состав серной кислоты
H2SO4 + 2H2O ↔ H3O+ * H2O

Функции кислотности
Теория Бренстеда – Лоури.
Согласно теории Бренстеда – Лоури кислота рассматривается
Функции кислотности
Теория Бренстеда – Лоури.
Согласно теории Бренстеда – Лоури кислота рассматривается

Физический смысл и меры основности в газовой фазе
Основностью в газовой
Физический смысл и меры основности в газовой фазе
Основностью в газовой

Протонирование слабых органических оснований в
водных растворах кислот.
Протонирование многих
Протонирование слабых органических оснований в
водных растворах кислот.
Протонирование многих

В зависимости от концентрации минеральной кислоты, характер специфической сольватации непрерывно меняется.
В зависимости от концентрации минеральной кислоты, характер специфической сольватации непрерывно меняется.

Гаммет и Дейруп: серия органических соединений, обладающие максимально сходной молекулярной структурой,
Гаммет и Дейруп: серия органических соединений, обладающие максимально сходной молекулярной структурой,
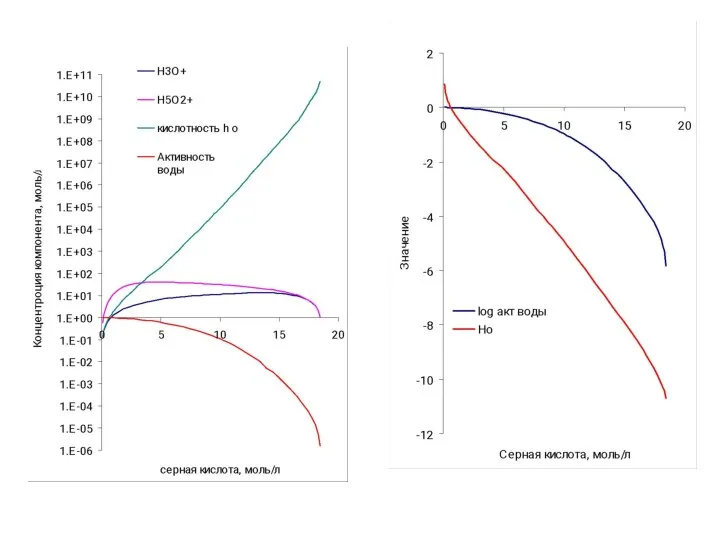
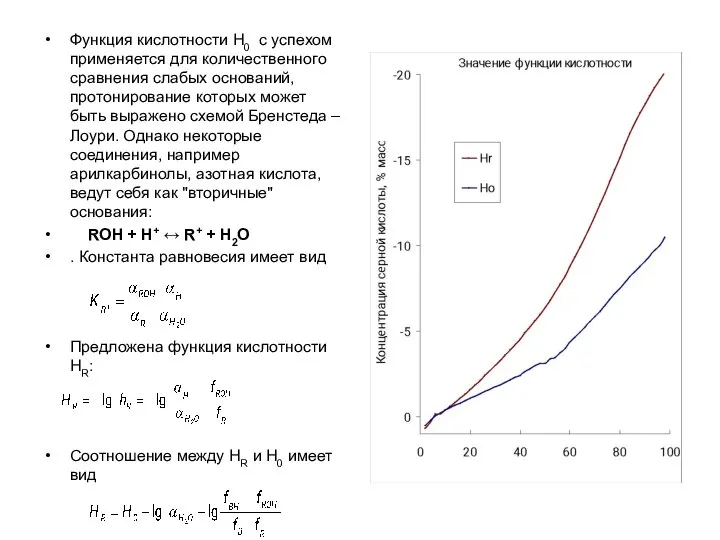
Функция кислотности Н0 с успехом применяется для количественного сравнения слабых оснований,
Функция кислотности Н0 с успехом применяется для количественного сравнения слабых оснований,
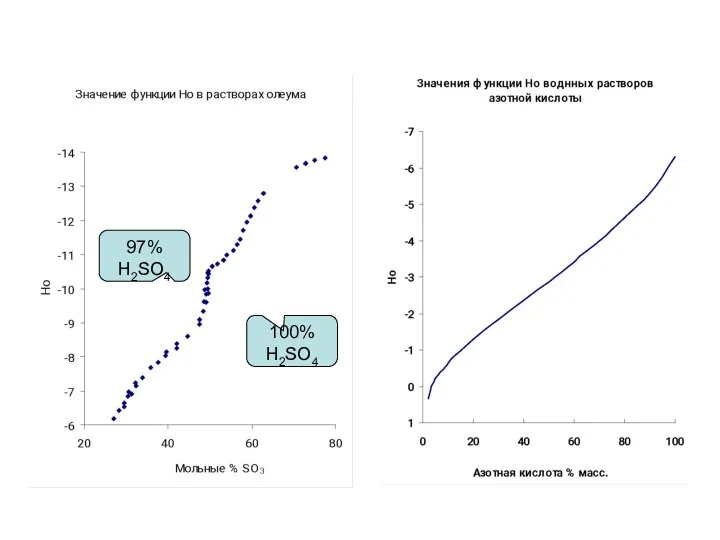
100% H2SO4
97% H2SO4
100% H2SO4
97% H2SO4

Кроме функций Нo и Hr существуют функции построенные на других рядах
Кроме функций Нo и Hr существуют функции построенные на других рядах
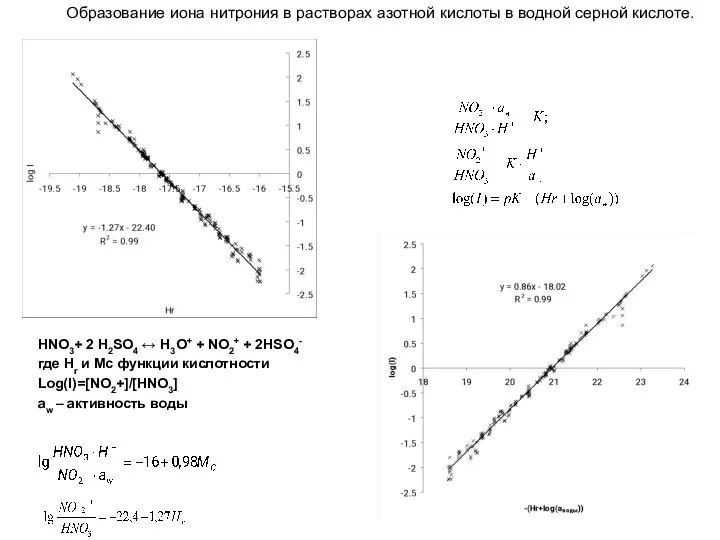
HNO3+ 2 Н2SO4 ↔ H3O+ + NO2+ + 2HSO4-
где Hr и
HNO3+ 2 Н2SO4 ↔ H3O+ + NO2+ + 2HSO4-
где Hr и
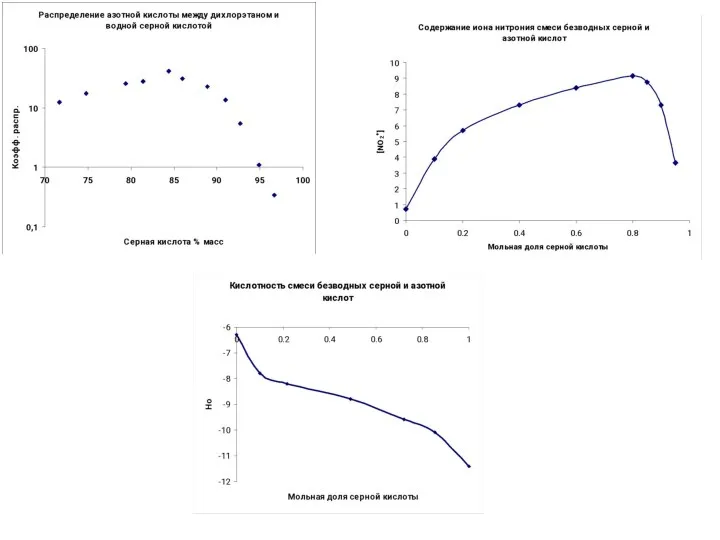

Лекция 3
Лекция 3
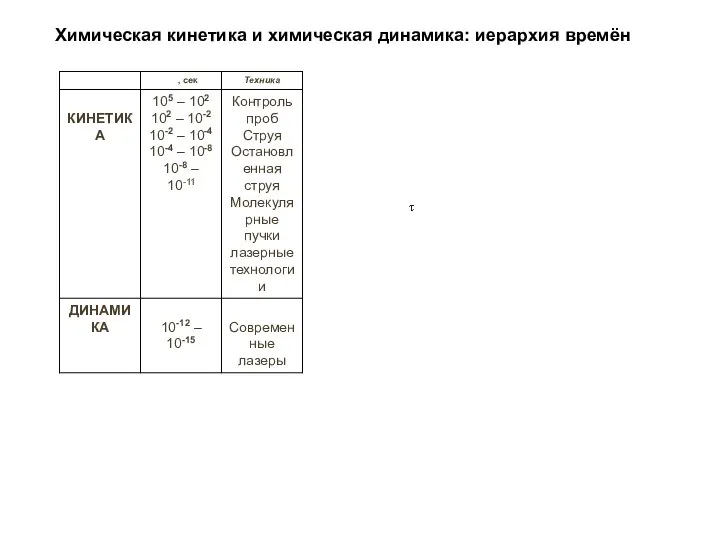
Химическая кинетика и химическая динамика: иерархия времён
Химическая кинетика и химическая динамика: иерархия времён
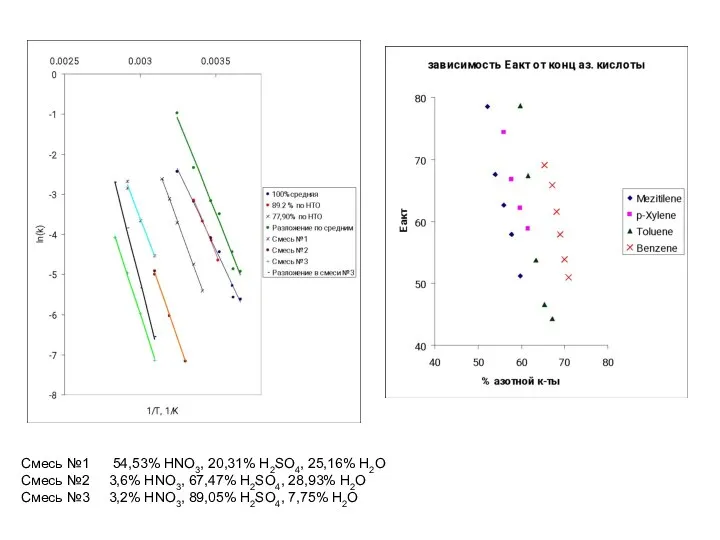
Смесь №1 54,53% HNO3, 20,31% H2SO4, 25,16% H2O
Смесь №2 3,6% HNO3,
Смесь №1 54,53% HNO3, 20,31% H2SO4, 25,16% H2O
Смесь №2 3,6% HNO3,

Пример кинетической кривой нитрования в серно-азотной нитрующей смеси, содержащей 73,5% серную
Пример кинетической кривой нитрования в серно-азотной нитрующей смеси, содержащей 73,5% серную
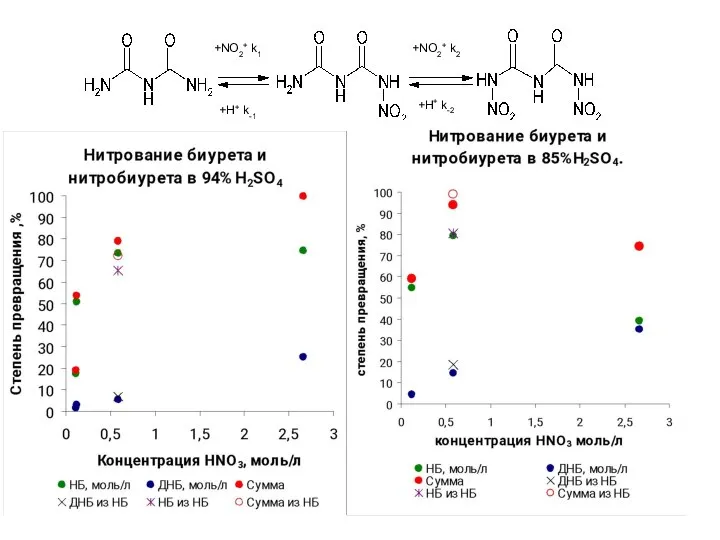
+NO2+ k1
+H+ k-1
+NO2+ k2
+H+ k-2
+NO2+ k1
+H+ k-1
+NO2+ k2
+H+ k-2
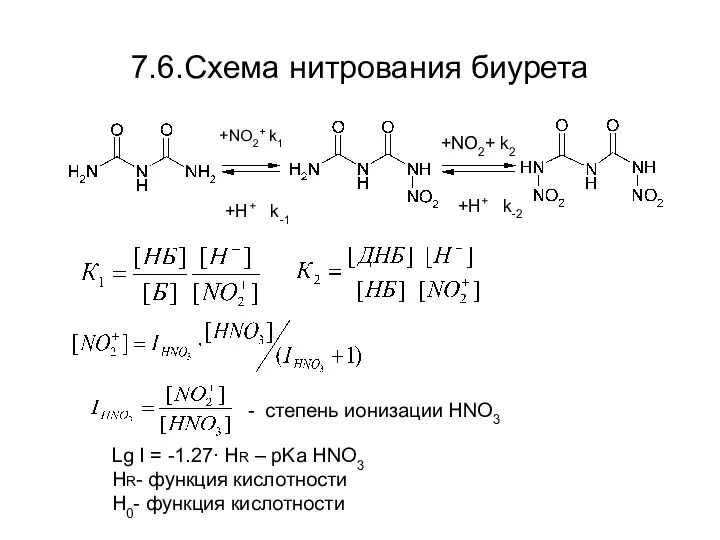
7.6.Схема нитрования биурета
+NO2+ k1
+H+ k-1
+NO2+ k2
+H+ k-2
- степень ионизации HNO3
Lg
7.6.Схема нитрования биурета
+NO2+ k1
+H+ k-1
+NO2+ k2
+H+ k-2
- степень ионизации HNO3
Lg

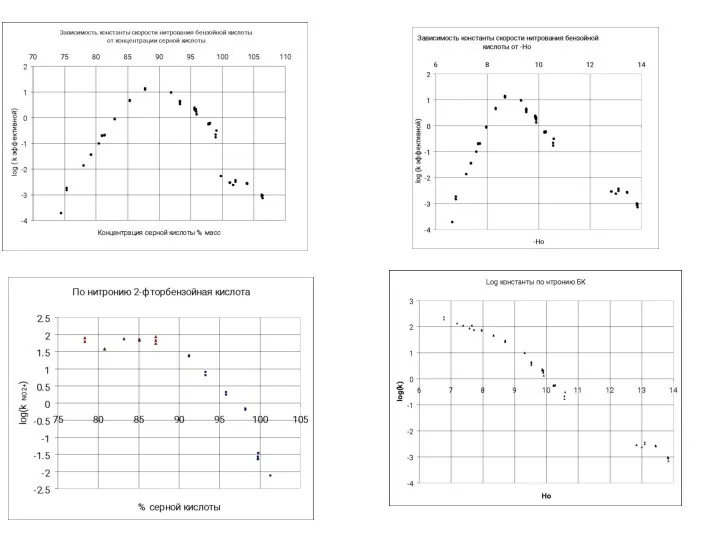
Примеры обработки кинетических данных по уравнению для последовательных реакций
80 %
85 %
93
Примеры обработки кинетических данных по уравнению для последовательных реакций
80 %
85 %
93
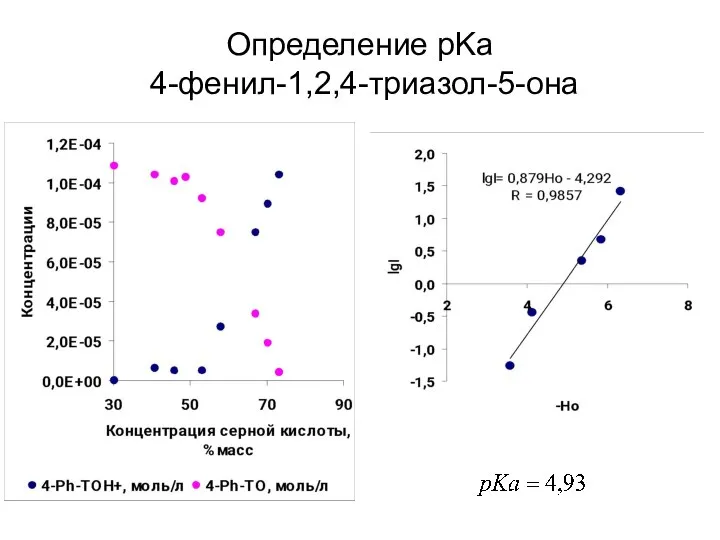
Определение pKa
4-фенил-1,2,4-триазол-5-она
Определение pKa
4-фенил-1,2,4-триазол-5-она
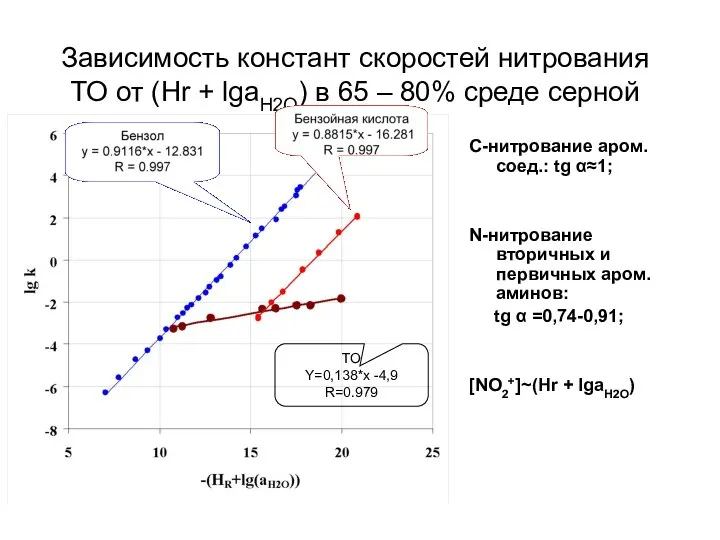
Зависимость констант скоростей нитрования ТО от (Hr + lgaH2O) в 65
Зависимость констант скоростей нитрования ТО от (Hr + lgaH2O) в 65
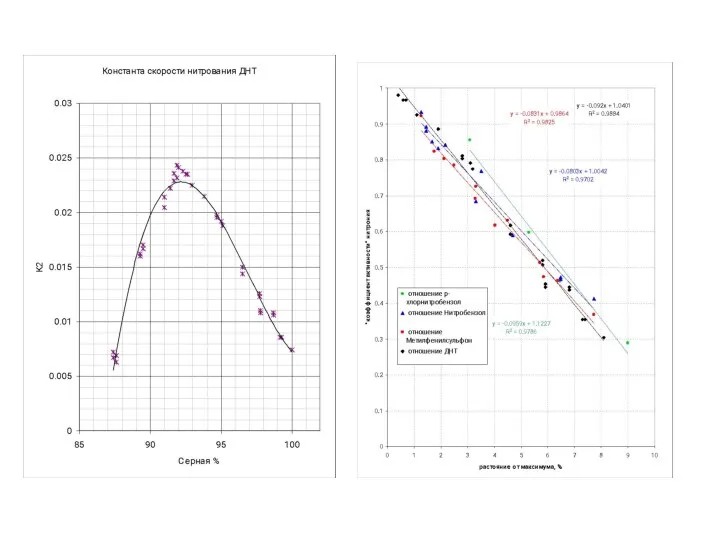
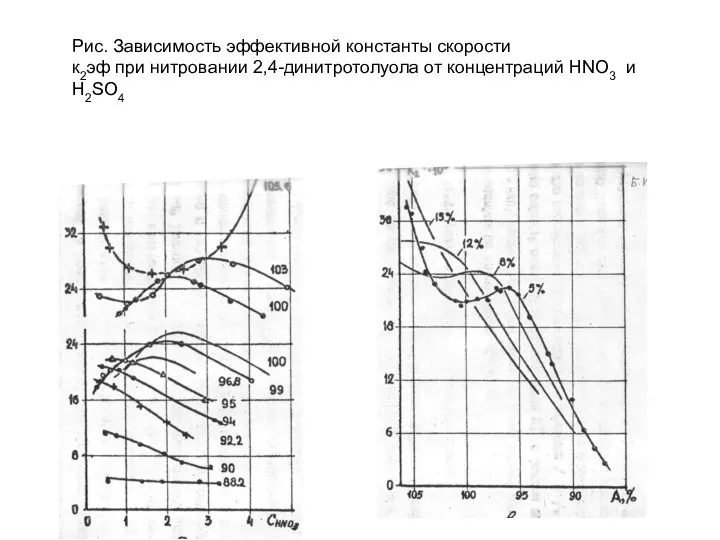
Рис. Зависимость эффективной константы скорости
к2эф при нитровании 2,4-динитротолуола от концентраций HNO3
Рис. Зависимость эффективной константы скорости
к2эф при нитровании 2,4-динитротолуола от концентраций HNO3

ТЕПЛОВЫЕ ЭФФЕКТЫ ПРИ НИТРОВАНИИ
Тепловой баланс.
Q приход = Q1 + Q2
ТЕПЛОВЫЕ ЭФФЕКТЫ ПРИ НИТРОВАНИИ
Тепловой баланс.
Q приход = Q1 + Q2

Суммарный тепловой эффект нитрования при получении
мононитротолуола ~ 123 кДж/мол (29
Суммарный тепловой эффект нитрования при получении
мононитротолуола ~ 123 кДж/мол (29
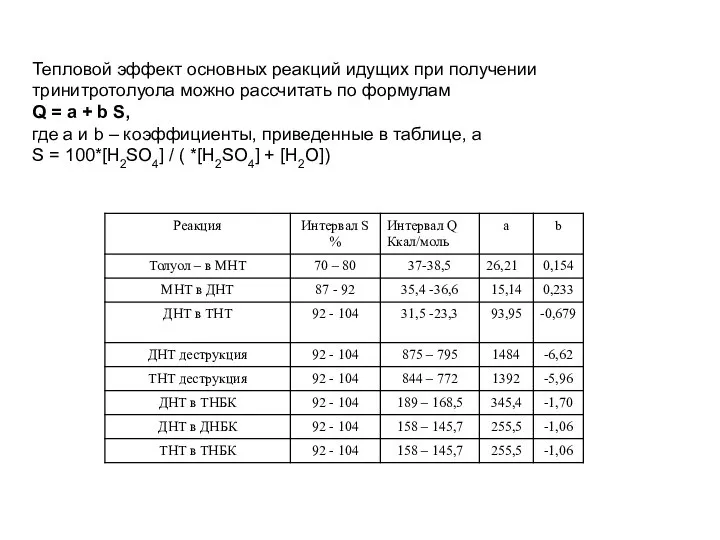
Тепловой эффект основных реакций идущих при получении тринитротолуола можно рассчитать по
Тепловой эффект основных реакций идущих при получении тринитротолуола можно рассчитать по

rD = β∑* (C2/γ –C1),
где β∑ -общее сопротивление диффузии
Если скорость
где β∑ -общее сопротивление диффузии
Если скорость
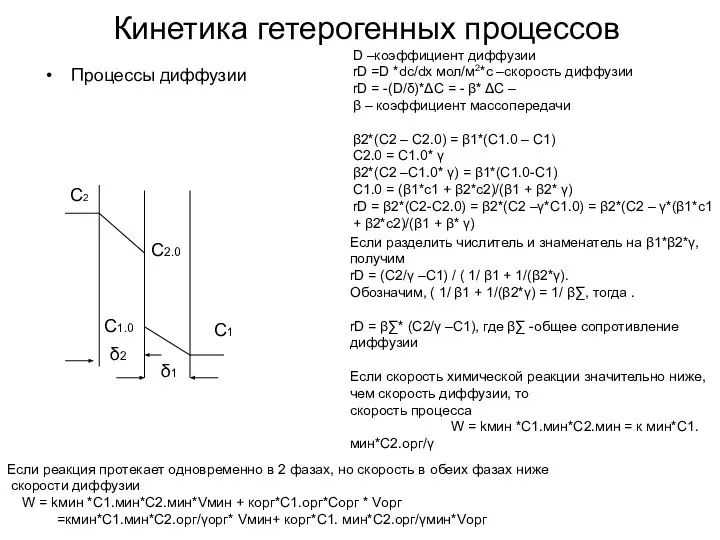
Кинетика гетерогенных процессов
Процессы диффузии
С2
С2.0
С1.0
С1
D –коэффициент диффузии
rD =D *dc/dx мол/м2*с –скорость диффузии
rD
Кинетика гетерогенных процессов
Процессы диффузии
С2
С2.0
С1.0
С1
D –коэффициент диффузии
rD =D *dc/dx мол/м2*с –скорость диффузии
rD

Влияние азотистой кислоты
В водных растворах H2SO4 и HClO4 (с
Влияние азотистой кислоты
В водных растворах H2SO4 и HClO4 (с

Изучение кинетики каталитического процесса строго доказало первоначальное нитрозирование с последующим быстрым
Изучение кинетики каталитического процесса строго доказало первоначальное нитрозирование с последующим быстрым

Последующие исследования показали, что при каталитическом действии низших оксидов азота
Последующие исследования показали, что при каталитическом действии низших оксидов азота

В 70-80-е годы образование относительно стабильных катион-радикалов (КР) спектральными методами или
В 70-80-е годы образование относительно стабильных катион-радикалов (КР) спектральными методами или
 Історичні відомості про спроби класифікації хімічних елементів. Відкриття періодичного закону Д.І. Менделєєва
Історичні відомості про спроби класифікації хімічних елементів. Відкриття періодичного закону Д.І. Менделєєва Карбоновые кислоты. Общая формула карбоновых кислот
Карбоновые кислоты. Общая формула карбоновых кислот Презентация по Химии "Утилизация попутного нефтяного газа" - скачать смотреть
Презентация по Химии "Утилизация попутного нефтяного газа" - скачать смотреть  Биохимия печени
Биохимия печени Переработка нефти. (10 класс)
Переработка нефти. (10 класс) Элементный, фракционный и химический состав нефти. Классификация нефтей
Элементный, фракционный и химический состав нефти. Классификация нефтей Отношение масс элементов в веществе. Массовые доли элементов в веществе
Отношение масс элементов в веществе. Массовые доли элементов в веществе Комплексні сполуки
Комплексні сполуки Презентация по Химии "Путешествие по стране карбонатов" - скачать смотреть
Презентация по Химии "Путешествие по стране карбонатов" - скачать смотреть  Вода в природе. Основные свойства воды
Вода в природе. Основные свойства воды Тест: Азот
Тест: Азот Органічна хімія
Органічна хімія Очистка белков (Разделение белков из гетерогенной белковой смеси)
Очистка белков (Разделение белков из гетерогенной белковой смеси) 1oe_zanyatie (1)
1oe_zanyatie (1) Физико-химические свойства лекарственных препаратов железа
Физико-химические свойства лекарственных препаратов железа Хлорид натрия (NaCI)
Хлорид натрия (NaCI) Строение атома. Периодичность свойств элементов и их соединений
Строение атома. Периодичность свойств элементов и их соединений Способы разделения смесей, применяемые в быту
Способы разделения смесей, применяемые в быту Химический состав клетки. Неорганические соединения
Химический состав клетки. Неорганические соединения Олово и свинец. (Лекция 2)
Олово и свинец. (Лекция 2) Проект познавательно-исследовательской деятельности. Удивительная соль
Проект познавательно-исследовательской деятельности. Удивительная соль Получение металлов
Получение металлов Предмет и задачи медтоксикологии
Предмет и задачи медтоксикологии Сложные эфиры. Жиры
Сложные эфиры. Жиры Жидкостная экстракция
Жидкостная экстракция Строение атома. Электронные оболочки атома
Строение атома. Электронные оболочки атома Обмен отдельных аминокислот. Образование аммиака и пути его обезвреживания в организме. Лекция 1
Обмен отдельных аминокислот. Образование аммиака и пути его обезвреживания в организме. Лекция 1 Комплексонометрическое титрование. 5 лекция. Часть 1
Комплексонометрическое титрование. 5 лекция. Часть 1