- Иммунодефициты

Содержание
- 2. Иммунный ответ
- 3. Микроорганизмы (1014) Постоянный пул в макроорганизме Проходящий пул Элиминация доиммунными механизмами Элиминация иммунными механизмами
- 4. Доиммунные механизмы Рецепторы Растворимые PRR (Pattern Recognition Receptors ) образ распознающие рецепторы 1. LPB ( Lipopolysaccharide
- 5. Toll-рецептор TNFα, IL-1,6,8,12 Костимуляция (CD-80, CD-86) Подготовка к адаптивному иммунитету
- 6. ИММУНОДЕФИЦИТЫ ПЕРВИЧНЫЕ (ПИД) Гуморальные Комбинированные Клеточные ВТОРИЧНЫЕ (ВИД) НАРУШЕНИЯ НЕСПЕЦИФИЧЕСКОЙ РЕЗИСТЕНТНОСТИ (врожденного иммунитета) Фагоцитоза Комплемента Toll-рецепторов
- 7. Первичные иммунодефициты – генетические или врожденные дефекты иммунной системы. Гуморальные или антительные с поражением В-лимфоцитов: Болезнь
- 8. Комбинированные иммунодефициты Синдром Вискотта –Олдрича Атаксия телеангиоэктозия (синдром Луи-Бар) Тяжелая комбинированная иммунная недостаточность Дефицит аденозиндезаминазы Дефицит
- 9. ВТОРИЧНЫЕ ИММУНОДЕФИЦИТЫ Дефекты питания Инфекции Гельминтозы Протеинурия при заболеваниях почек Хроническая почечная недостаточность Диарея Стресс Эндокринопатии
- 10. Первичные иммунодефициты
- 11. Гуморальные иммунодефициты
- 12. Болезнь Брутона (Х-сцепленная агаммаглобулинемия) Мутация гена btk, участвующего в синтезе иммуноглобулинов и в созревании В-лимфоцитов Лечение:
- 13. Селективный дефицит IgA Связан с повреждением 18 хромосомы Происходит нарушение нормальной последовательность синтеза Ig плазмоцитами IgM
- 14. Селективный дефицит подклассов IgG Наиболее часто происходит снижение подкласса IgG2, общий IgG при этом остается в
- 15. Гипер-IgM-иммунодефицит Связан с повреждением гена CD40L Повышено содержание IgM Снижено содержание IgG и IgA Происходит нарушение
- 16. Транзиторная гипогаммаглобулинемия новорожденных Такой ИД можно считать физиологическим Причина – недозревание Т и В-лимфоцитов ребенка Первые
- 17. Комбинированные иммунодефициты
- 18. Тяжелый комбинированный иммунодефицит (ТКИД) Х-сцепленный ТКИД ТКИД с аутосомно-рецессивным наследованием
- 19. Х-сцепленный ТКИД Связан с повреждением гена Xq13 ответственного за синтез рецепторов на лимфоцитах для интерлейкинов: IL-2,
- 20. ТКИД с аутосомно-рецессивным наследованием Связан с дефицитом аденозиндезаминазы (АДА) (поражен ген 20q-13-ter) В результате дефицита АДА
- 21. Синдром Луи-Барр (атаксия-телеангиоэктазия) (поражен ген 11q-22-23, контролирующий клеточные митозы и синтез рецепторов для митогенных сигналов) В
- 22. Синдром Вискотта-Олдрича Связан с поражением гена кодирующего группу WASP-белков, обеспечивающих синтез внутриклеточного актина (цитоскелета). В результате
- 23. Общий вариабельный иммунодефицит Связан с повреждением гена CD40L Проявляется на поздних этапах онтогенеза в 25 –
- 24. Клеточные иммунодефициты
- 25. Синдром Ди Джорджи Не наследственный а врожденный, связанный с нарушением эмбриогенеза в момент закладки III и
- 26. Нарушения неспецифической резистентности
- 27. Хроническая гранулематозная болезнь Генетический дефект НАДФ-оксидазы, что приводит к дефициту супероксид-аниона. В результате фагоцитоз идет, а
- 28. Синдром Чедиак-Хигаси Лизосомы фагоцита не способны сливаться с фагосомой, секрет в них накапливается и образуются гигантские
- 29. Дефицит системы комплемента – Врожденный ангионевротический отек – отек Квинке Обусловлен дефицитом С1-inh – ингибитора системы
- 30. Вторичные иммунодефициты
- 31. ВИД при стрессе Повышенная выработка ГКС как реакция на стресс Снижение размножения и синтеза белков кортизон-чувствительных
- 32. ВИД как осложнение иммуносупрессивной терапии Для иммуносупрессивной терапии применяются цитостатические препараты, подавляющие иммунный ответ в результате
- 33. ВИД у пожилых людей Возможное объяснение – «ампутация клонов лимфоцитов» - истощение запасов стволовых клеток при
- 34. ВИД при ожоговой болезни Связан с массивной потерей белка с плазмой через ожоговую поверхность Потеря источника
- 35. ВИД при лучевой болезни Ионизирующая радиация обладает цитостатическим влиянием, в результате чего к симптомам лучевого поражения
- 36. ВИД при голодании Необходимость постоянного размножения иммунокомпетентных клеток делает иммунную систему уязвимой к дефициту белков и
- 37. ВИД при дефиците витамина В12 и фолиевой кислоты Данные витамины необходимы для синтеза пуриновых оснований –
- 38. ВИД при дефиците железа Ион железа входит в состав фермента макрофагов миелопероксидазы необходимого для лизиса сфагоцитированных
- 39. ВИД после хирургических вмешательств Хирургическое вмешательство может вызвать стресс и иммунодепрессию
- 40. ВИД при беременности Является физиологическим состоянием, необходим для вынашивания чужеродного для беременной по антигенному составу организма
- 42. Скачать презентацию
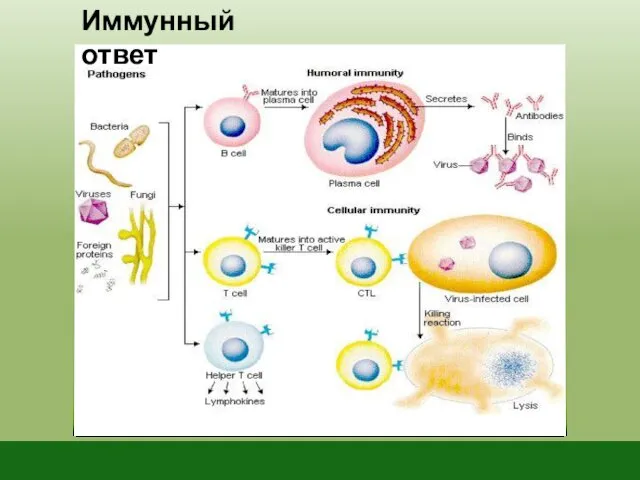
Иммунный ответ
Иммунный ответ
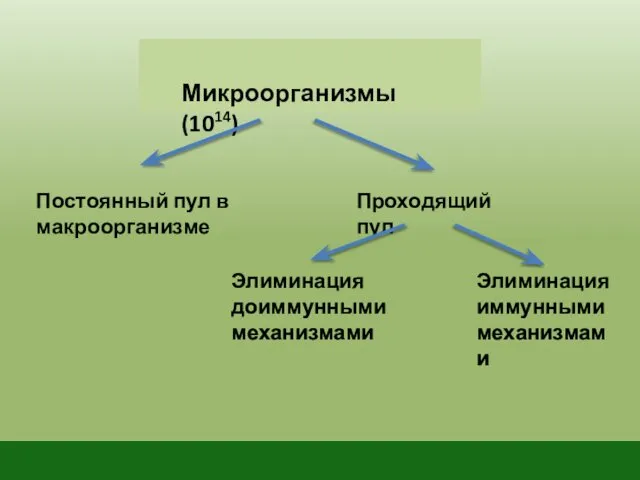
Микроорганизмы (1014)
Постоянный пул в макроорганизме
Проходящий пул
Элиминация доиммунными механизмами
Элиминация иммунными механизмами
Микроорганизмы (1014)
Постоянный пул в макроорганизме
Проходящий пул
Элиминация доиммунными механизмами
Элиминация иммунными механизмами

Доиммунные механизмы
Рецепторы
Растворимые PRR (Pattern Recognition Receptors ) образ распознающие рецепторы
1.
Доиммунные механизмы
Рецепторы
Растворимые PRR (Pattern Recognition Receptors ) образ распознающие рецепторы
1.
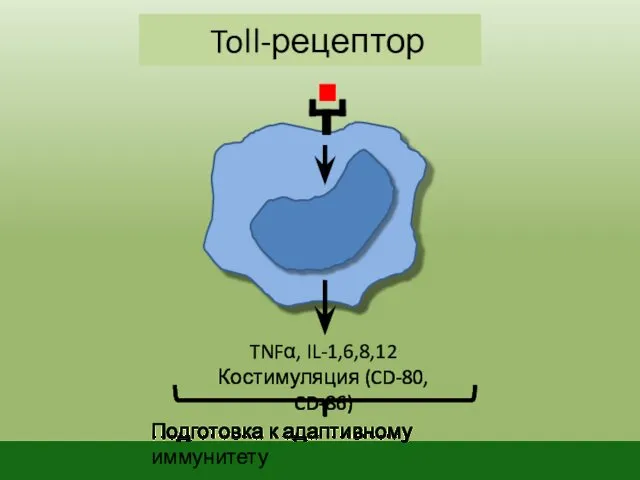
Toll-рецептор
TNFα, IL-1,6,8,12
Костимуляция (CD-80, CD-86)
Подготовка к адаптивному иммунитету
Toll-рецептор
TNFα, IL-1,6,8,12
Костимуляция (CD-80, CD-86)
Подготовка к адаптивному иммунитету
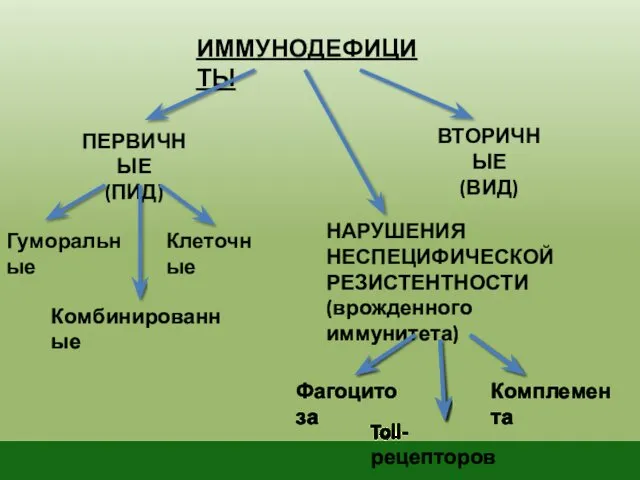
ИММУНОДЕФИЦИТЫ
ПЕРВИЧНЫЕ
(ПИД)
Гуморальные
Комбинированные
Клеточные
ВТОРИЧНЫЕ
(ВИД)
НАРУШЕНИЯ НЕСПЕЦИФИЧЕСКОЙ РЕЗИСТЕНТНОСТИ
(врожденного иммунитета)
Фагоцитоза
Комплемента
Toll-рецепторов
ИММУНОДЕФИЦИТЫ
ПЕРВИЧНЫЕ
(ПИД)
Гуморальные
Комбинированные
Клеточные
ВТОРИЧНЫЕ
(ВИД)
НАРУШЕНИЯ НЕСПЕЦИФИЧЕСКОЙ РЕЗИСТЕНТНОСТИ
(врожденного иммунитета)
Фагоцитоза
Комплемента
Toll-рецепторов

Первичные иммунодефициты – генетические или врожденные дефекты иммунной системы.
Гуморальные
Первичные иммунодефициты – генетические или врожденные дефекты иммунной системы.
Гуморальные

Комбинированные иммунодефициты
Синдром Вискотта –Олдрича
Атаксия телеангиоэктозия (синдром Луи-Бар)
Тяжелая комбинированная иммунная недостаточность
Дефицит
Комбинированные иммунодефициты
Синдром Вискотта –Олдрича
Атаксия телеангиоэктозия (синдром Луи-Бар)
Тяжелая комбинированная иммунная недостаточность
Дефицит

ВТОРИЧНЫЕ ИММУНОДЕФИЦИТЫ
Дефекты питания
Инфекции
Гельминтозы
Протеинурия при заболеваниях почек
Хроническая почечная недостаточность
Диарея
Стресс
Эндокринопатии
Лекарства ( глюкокортикоиды, цитостатики,
ВТОРИЧНЫЕ ИММУНОДЕФИЦИТЫ
Дефекты питания
Инфекции
Гельминтозы
Протеинурия при заболеваниях почек
Хроническая почечная недостаточность
Диарея
Стресс
Эндокринопатии
Лекарства ( глюкокортикоиды, цитостатики,

Первичные иммунодефициты
Первичные иммунодефициты

Гуморальные иммунодефициты
Гуморальные иммунодефициты

Болезнь Брутона
(Х-сцепленная агаммаглобулинемия)
Мутация гена btk, участвующего в синтезе иммуноглобулинов и в
Болезнь Брутона
(Х-сцепленная агаммаглобулинемия)
Мутация гена btk, участвующего в синтезе иммуноглобулинов и в

Селективный дефицит IgA
Связан с повреждением 18 хромосомы
Происходит нарушение нормальной последовательность
Селективный дефицит IgA
Связан с повреждением 18 хромосомы
Происходит нарушение нормальной последовательность

Селективный дефицит подклассов IgG
Наиболее часто происходит снижение подкласса IgG2, общий IgG
Селективный дефицит подклассов IgG
Наиболее часто происходит снижение подкласса IgG2, общий IgG

Гипер-IgM-иммунодефицит
Связан с повреждением гена CD40L
Повышено содержание IgM
Снижено содержание IgG и IgA
Гипер-IgM-иммунодефицит
Связан с повреждением гена CD40L
Повышено содержание IgM
Снижено содержание IgG и IgA

Транзиторная гипогаммаглобулинемия новорожденных
Такой ИД можно считать физиологическим
Причина – недозревание Т и
Транзиторная гипогаммаглобулинемия новорожденных
Такой ИД можно считать физиологическим
Причина – недозревание Т и

Комбинированные
иммунодефициты
Комбинированные
иммунодефициты

Тяжелый комбинированный иммунодефицит (ТКИД)
Х-сцепленный ТКИД
ТКИД с аутосомно-рецессивным
наследованием
Тяжелый комбинированный иммунодефицит (ТКИД)
Х-сцепленный ТКИД
ТКИД с аутосомно-рецессивным
наследованием

Х-сцепленный ТКИД
Связан с повреждением гена Xq13
ответственного за синтез рецепторов
Х-сцепленный ТКИД
Связан с повреждением гена Xq13
ответственного за синтез рецепторов

ТКИД с аутосомно-рецессивным
наследованием
Связан с дефицитом аденозиндезаминазы (АДА)
(поражен ген 20q-13-ter)
В
ТКИД с аутосомно-рецессивным
наследованием
Связан с дефицитом аденозиндезаминазы (АДА)
(поражен ген 20q-13-ter)
В
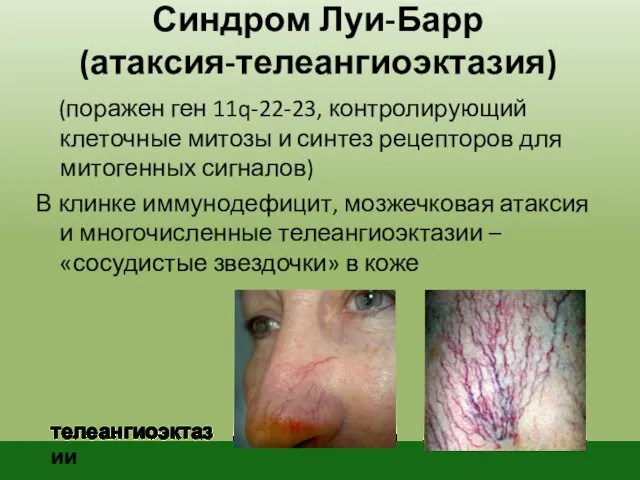
Синдром Луи-Барр
(атаксия-телеангиоэктазия)
(поражен ген 11q-22-23, контролирующий клеточные митозы и синтез рецепторов
Синдром Луи-Барр
(атаксия-телеангиоэктазия)
(поражен ген 11q-22-23, контролирующий клеточные митозы и синтез рецепторов

Синдром Вискотта-Олдрича
Связан с поражением гена кодирующего группу WASP-белков, обеспечивающих синтез внутриклеточного
Синдром Вискотта-Олдрича
Связан с поражением гена кодирующего группу WASP-белков, обеспечивающих синтез внутриклеточного

Общий вариабельный иммунодефицит
Связан с повреждением гена CD40L
Проявляется на поздних этапах онтогенеза
Общий вариабельный иммунодефицит
Связан с повреждением гена CD40L
Проявляется на поздних этапах онтогенеза

Клеточные
иммунодефициты
Клеточные
иммунодефициты

Синдром Ди Джорджи
Не наследственный а врожденный, связанный с нарушением эмбриогенеза в
Синдром Ди Джорджи
Не наследственный а врожденный, связанный с нарушением эмбриогенеза в

Нарушения неспецифической резистентности
Нарушения неспецифической резистентности
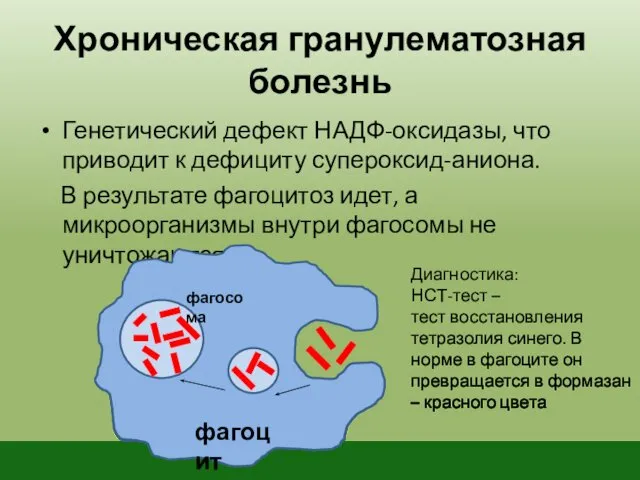
Хроническая гранулематозная болезнь
Генетический дефект НАДФ-оксидазы, что приводит к дефициту супероксид-аниона.
Хроническая гранулематозная болезнь
Генетический дефект НАДФ-оксидазы, что приводит к дефициту супероксид-аниона.

Синдром Чедиак-Хигаси
Лизосомы фагоцита не
способны сливаться с
фагосомой, секрет в них
Синдром Чедиак-Хигаси
Лизосомы фагоцита не
способны сливаться с
фагосомой, секрет в них
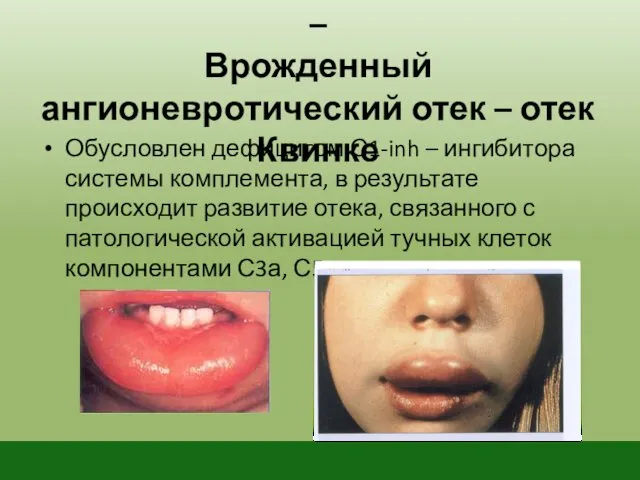
Дефицит системы комплемента –
Врожденный ангионевротический отек – отек Квинке
Обусловлен дефицитом
Дефицит системы комплемента –
Врожденный ангионевротический отек – отек Квинке
Обусловлен дефицитом

Вторичные иммунодефициты
Вторичные иммунодефициты

ВИД при стрессе
Повышенная выработка ГКС как реакция на стресс
Снижение размножения и
ВИД при стрессе
Повышенная выработка ГКС как реакция на стресс
Снижение размножения и

ВИД как осложнение иммуносупрессивной терапии
Для иммуносупрессивной терапии применяются цитостатические препараты, подавляющие
ВИД как осложнение иммуносупрессивной терапии
Для иммуносупрессивной терапии применяются цитостатические препараты, подавляющие

ВИД у пожилых людей
Возможное объяснение – «ампутация клонов лимфоцитов» - истощение
ВИД у пожилых людей
Возможное объяснение – «ампутация клонов лимфоцитов» - истощение

ВИД при ожоговой болезни
Связан с массивной потерей белка с плазмой через
ВИД при ожоговой болезни
Связан с массивной потерей белка с плазмой через

ВИД при лучевой болезни
Ионизирующая радиация обладает
цитостатическим влиянием, в результате чего
ВИД при лучевой болезни
Ионизирующая радиация обладает
цитостатическим влиянием, в результате чего

ВИД при голодании
Необходимость постоянного размножения иммунокомпетентных клеток делает иммунную систему уязвимой
ВИД при голодании
Необходимость постоянного размножения иммунокомпетентных клеток делает иммунную систему уязвимой

ВИД при дефиците витамина В12 и фолиевой кислоты
Данные витамины необходимы для
ВИД при дефиците витамина В12 и фолиевой кислоты
Данные витамины необходимы для

ВИД при дефиците железа
Ион железа входит в состав фермента макрофагов миелопероксидазы
ВИД при дефиците железа
Ион железа входит в состав фермента макрофагов миелопероксидазы

ВИД после хирургических вмешательств
Хирургическое вмешательство может вызвать стресс и иммунодепрессию
ВИД после хирургических вмешательств
Хирургическое вмешательство может вызвать стресс и иммунодепрессию

ВИД при беременности
Является физиологическим состоянием, необходим для вынашивания чужеродного для беременной
ВИД при беременности
Является физиологическим состоянием, необходим для вынашивания чужеродного для беременной
 Особенности сестринской деятельности при уходе за пациентами с мочекаменной болезнью на стационарном этапе
Особенности сестринской деятельности при уходе за пациентами с мочекаменной болезнью на стационарном этапе Узкий таз в современном акушерстве
Узкий таз в современном акушерстве Профессиональные заболевания медицинских работников, обусловленные перенапряжением опорно-двигательного аппарата
Профессиональные заболевания медицинских работников, обусловленные перенапряжением опорно-двигательного аппарата Oбщeниe в сестринском деле
Oбщeниe в сестринском деле Правила безопасного сексуального поведения
Правила безопасного сексуального поведения Учение о критических периодах развития человека
Учение о критических периодах развития человека Акарозы птиц. Цикл развития куриного клеща
Акарозы птиц. Цикл развития куриного клеща Эпидемиологическая ситуация по туберкулезу среди детей в Пермском крае
Эпидемиологическая ситуация по туберкулезу среди детей в Пермском крае Черепно–мозговая травма
Черепно–мозговая травма Современные аспекты лечения болезни Паркинсона
Современные аспекты лечения болезни Паркинсона Методы диагностики при заболевании сердечно-сосудистой системы
Методы диагностики при заболевании сердечно-сосудистой системы Пахова грижа у дітей
Пахова грижа у дітей Профессиональная гигиена полости рта
Профессиональная гигиена полости рта Рак гортани
Рак гортани Возрастная физиология
Возрастная физиология Строение зубов
Строение зубов Перитонеальный диализ
Перитонеальный диализ Невропатия лучевого, локтевого и срединного нервов
Невропатия лучевого, локтевого и срединного нервов Электронды базалардан артық салмақ және қант диабеті арасындағы байланыс жайлы мәліметтерді ғылыми статьялардан табу
Электронды базалардан артық салмақ және қант диабеті арасындағы байланыс жайлы мәліметтерді ғылыми статьялардан табу Оптическое излучение
Оптическое излучение Үлкен жартышар туралы қысқаша мәлімет
Үлкен жартышар туралы қысқаша мәлімет Чувствительная сфера человека
Чувствительная сфера человека Правовые основы оказания первой помощи. Принципы, этапы, мероприятия оказания первой помощи
Правовые основы оказания первой помощи. Принципы, этапы, мероприятия оказания первой помощи Амобиаз
Амобиаз Плеврит. Классификация
Плеврит. Классификация Ішкі аурулар. Жүрек-тамыр жүйесі
Ішкі аурулар. Жүрек-тамыр жүйесі Острая массивная кровопотеря
Острая массивная кровопотеря Острые кишечные инфекции у взрослых и детей
Острые кишечные инфекции у взрослых и детей