- Митохондриальный геном и болезни человека

Содержание
- 2. 1- хп ячмень (ПК) 2- митох ячмень (КМ) 3- хп ячмень (СвЦ) 4 - хп ячмень
- 3. Митохондрии в клетке, строение митохондрий
- 4. Функции митохондрий Синтез АТФ – «энергетический центр» клетки (95% синтезируется в мт) Участие в метаболизме аминокислот,
- 5. Митохондриальный геном человека – 37 генов, 16. 569 пар нуклеотидов 2 гена рибосомальной РНК 22 гена
- 6. Митохондриальный геном человека
- 7. Репликация митохондриальной ДНК млекопитающих L-цепь Н-цепь новая Н-цепь (7S ДНК) D-петля мтДНК ОL ОH ОH HSP
- 8. ДНК- полимераза гамма(γ) состоит из двух субъединиц: α - каталитической и β -дополнительной (accessory) 125 -140
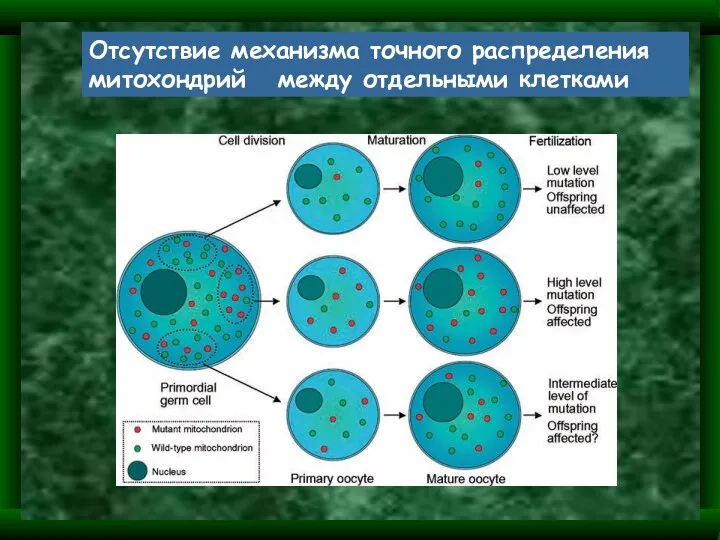
- 10. Особености митохондриальной наследственности Материнское наследование Мультикопийность геномов (сотни органелл, тысячи ДНК молекул) Гетероплазмия Митотическая сегрегация Пороговый
- 11. Ткани с низким порогом мутантной ДНК: мозг сердце скелетная мускулатура сетчатка глаза почечные канальцы эндокринные железы
- 13. Мутации в митохондриальной ДНК человека происходят в пять раз чаще, чем в ядерной, поскольку геном митохондрий
- 14. В настоящее время описано более 190 патогенных точечных мутаций митохондриальной ДНК около 200 делеций, инсерций и
- 15. Источники митохондриальных патологий: Изменения в генах ядерного кодирования (более 1000 генов кодируют мт белки) Изменения в
- 16. Особенности мутаций митохондриальных генов Одна из важнейших особенностей – клиническое разнообразие сиблингов. Это – отражение «эффекта
- 18. Уменьшение синтеза АТФ Нарушение кальциевого баланса клетки Повышение количества ROS (reactive oxygen species) Особенности мутаций митохондриальных
- 19. ген ДНК-полимеразы гамма (осуществляет синтез мтДНК); ген тимидинфосфорилазы (нарушает метаболизм тимидина); ген Twinkle (участвует в поддержании
- 20. Особенности мутации митохондриальных генов Строгая связь между сайтом мутации и клиническим фенотипом часто отсутствует: Одна и
- 21. 2/3 известных точечных мутаций мтДНК сосредоточены в тРНК генах (9% генома) Больше всего мутантных точек выявлено
- 22. Мутации лейциновой тРНК
- 23. Синдром Лебера: LHON (1871 г.) наследуемая по материнской линии потеря зрения происходит у людей 20-30 лет
- 24. ДНК-диагностика синдрома Лебера в семье N проведена нами впервые в 2006 году здоровый сестра мать человек
- 25. Лишь у 50% мужчин и 10% женщин носителей патогенных мутаций комплекса I в действительности происходит потеря
- 26. Мутации генов транспортной РНК Самая часто встречающаяся точечная мутация: А3243G в лейциновой тРНК Обнаружена у большинства
- 27. Впервые ДНК-диагностика синдрома MELAS была проведена нами в 2007 году Мама: фенотипически здоровая женщина очень маленького
- 28. Мутация резко снижает эффективность трансляции в мт и тем самым провоцирует дефицит дыхательной цепи
- 29. Мутации генов рибосомальной РНК Чаще всего встречается мутация гена 12S рРНК A1555G Вызывает несиндромную потерю слуха
- 30. T8993G: лейцин замещается на аргинин в ATPase6, что приводит к нарушению синтеза АТФ Если доля мтДНК
- 31. Синдром Лея – тяжелейшее нейродегенеративное заболевание: - симметричные некротические повреждения в субкортикальных областях ЦНС – базальных
- 32. LS 2/10 случаев – мутации митохондр. ДНК (MILS) 1/10 cлучаев – мутации Х-хромосомы ATPase 6 PDHC
- 34. Та же делеция 5 т.п.н. вызывает еще 2 синдрома: Синдром PEO – Прогрессирующая наружная офтальмоплегия Синдром
- 35. В случае этой же делеции в 5 тыс п.н. вместо фатального KSS может наблюдаться PEO Прогрессирующая
- 36. Синдром митохондриальной деплеции - МDS В клетках остается 1 - 30% от нормального количества мтДНК Синдром
- 37. Патологии, вызванные изменением генов дыхательной цепи LHON LHON+дистония Спорадическая миопатия Спорадическая миопатия Спорадическая миопатия Энцефаломиопатия NARP
- 46. Другой подход - уменьшить соотношение мутантная:нормальная мтДНК I. Увеличить количество немутантных молекул путем «сдвига генов» Индуцируется
- 47. II.Уменьшить количество мутантных молекул мтДНК Разработка синтетических молекул, избирательно связывающихся с мутаными ДНК и блокирующих их
- 48. Лечение митохондриальных болезней (4) Импорт из цитоплазмы нормальных тРНК вместо дефектных митохондриальных Замена дефектного комплекса дых.
- 49. Лечение митохондриальных болезней –насколько это реально? Фармакологическое Вылечить от митохондриального заболевания сегодня невозможно Применяется симптоматическое лечение:
- 50. Ряд препаратов провоцирует митохондриальные заболевания или отягощает их течение Вальпроат: увеличивает частоту судорог при MELAS, гепатотоксичен
- 52. Скачать презентацию
1- хп ячмень (ПК)
2- митох ячмень (КМ)
3- хп ячмень (СвЦ)
4 -
1- хп ячмень (ПК)
2- митох ячмень (КМ)
3- хп ячмень (СвЦ)
4 -
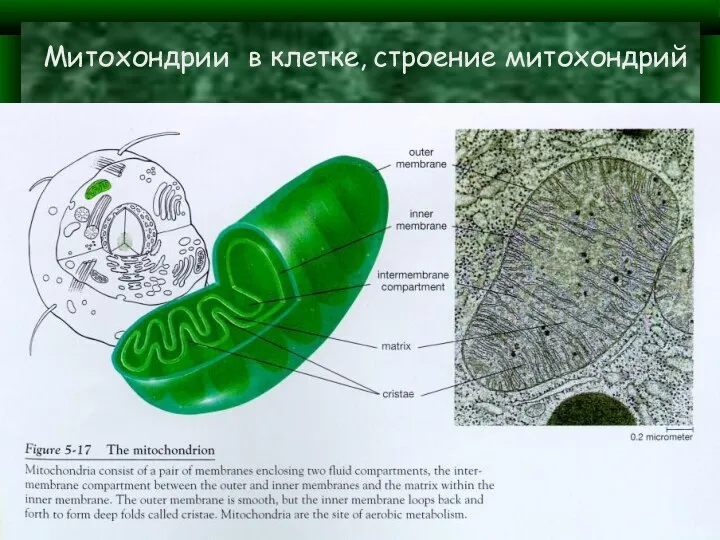
Митохондрии в клетке, строение митохондрий
Митохондрии в клетке, строение митохондрий

Функции митохондрий
Синтез АТФ – «энергетический центр» клетки (95% синтезируется в мт)
Участие
Функции митохондрий
Синтез АТФ – «энергетический центр» клетки (95% синтезируется в мт)
Участие

Митохондриальный геном человека
– 37 генов, 16. 569 пар нуклеотидов
2 гена
Митохондриальный геном человека
– 37 генов, 16. 569 пар нуклеотидов
2 гена
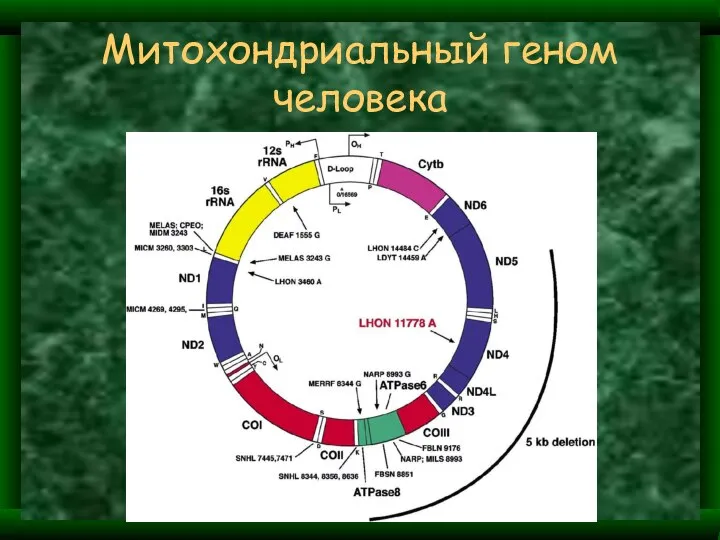
Митохондриальный геном человека
Митохондриальный геном человека
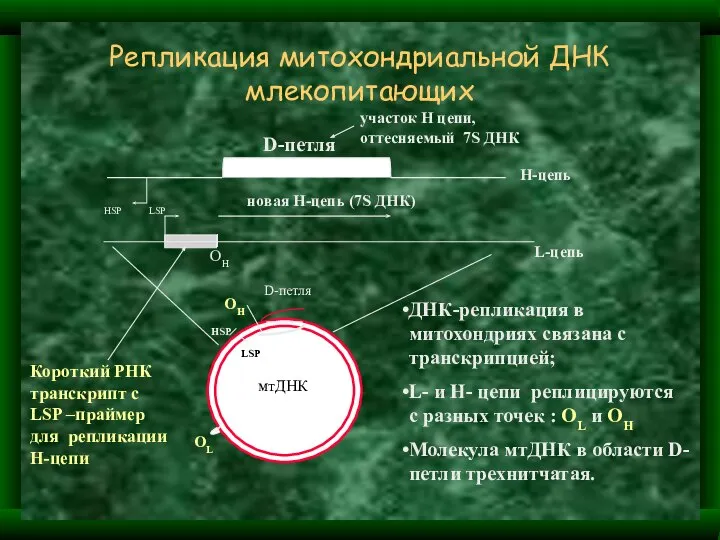
Репликация митохондриальной ДНК млекопитающих
L-цепь
Н-цепь
новая Н-цепь (7S ДНК)
D-петля
мтДНК
ОL
ОH
ОH
HSP LSP
Репликация митохондриальной ДНК млекопитающих
L-цепь
Н-цепь
новая Н-цепь (7S ДНК)
D-петля
мтДНК
ОL
ОH
ОH
HSP LSP

ДНК- полимераза гамма(γ)
состоит из двух субъединиц:
α - каталитической и
ДНК- полимераза гамма(γ)
состоит из двух субъединиц:
α - каталитической и


Особености митохондриальной наследственности
Материнское наследование
Мультикопийность геномов (сотни органелл, тысячи ДНК молекул)
Гетероплазмия
Митотическая сегрегация
Пороговый
Особености митохондриальной наследственности
Материнское наследование
Мультикопийность геномов (сотни органелл, тысячи ДНК молекул)
Гетероплазмия
Митотическая сегрегация
Пороговый

Ткани с низким порогом мутантной ДНК:
мозг
сердце
скелетная мускулатура
сетчатка глаза
почечные канальцы
эндокринные железы
Клетки этих
Ткани с низким порогом мутантной ДНК:
мозг
сердце
скелетная мускулатура
сетчатка глаза
почечные канальцы
эндокринные железы
Клетки этих


Мутации в митохондриальной ДНК
человека происходят в пять раз чаще,
чем
Мутации в митохондриальной ДНК
человека происходят в пять раз чаще,
чем

В настоящее время описано
более 190 патогенных точечных мутаций митохондриальной
В настоящее время описано
более 190 патогенных точечных мутаций митохондриальной

Источники митохондриальных патологий:
Изменения в генах ядерного кодирования
(более 1000 генов
Источники митохондриальных патологий:
Изменения в генах ядерного кодирования
(более 1000 генов
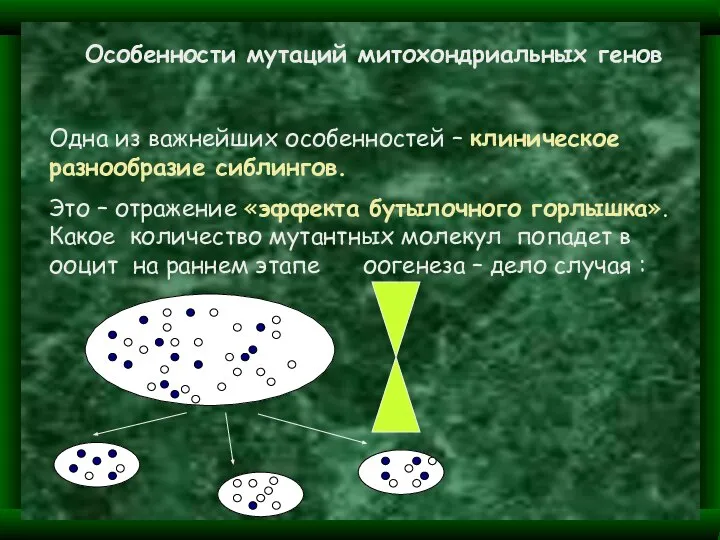
Особенности мутаций митохондриальных генов
Одна из важнейших особенностей – клиническое разнообразие
Особенности мутаций митохондриальных генов
Одна из важнейших особенностей – клиническое разнообразие

Уменьшение синтеза АТФ
Нарушение кальциевого баланса клетки
Повышение количества ROS (reactive oxygen species)
Особенности
Уменьшение синтеза АТФ
Нарушение кальциевого баланса клетки
Повышение количества ROS (reactive oxygen species)
Особенности

ген ДНК-полимеразы гамма
(осуществляет синтез мтДНК);
ген
ген ДНК-полимеразы гамма
(осуществляет синтез мтДНК);
ген
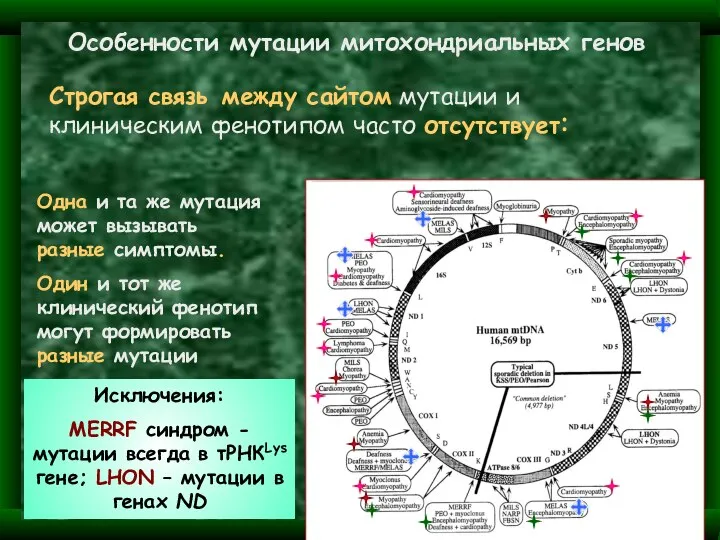
Особенности мутации митохондриальных генов
Строгая связь между сайтом мутации и клиническим фенотипом
Особенности мутации митохондриальных генов
Строгая связь между сайтом мутации и клиническим фенотипом

2/3 известных точечных мутаций
мтДНК сосредоточены
в тРНК генах (9% генома)
Больше всего
2/3 известных точечных мутаций
мтДНК сосредоточены
в тРНК генах (9% генома)
Больше всего
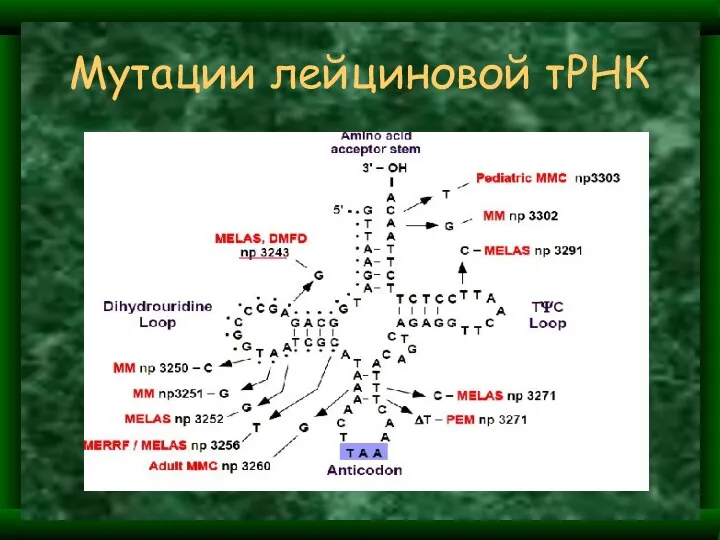
Мутации лейциновой тРНК
Мутации лейциновой тРНК

Синдром Лебера: LHON (1871 г.)
наследуемая по материнской линии потеря зрения
Синдром Лебера: LHON (1871 г.)
наследуемая по материнской линии потеря зрения
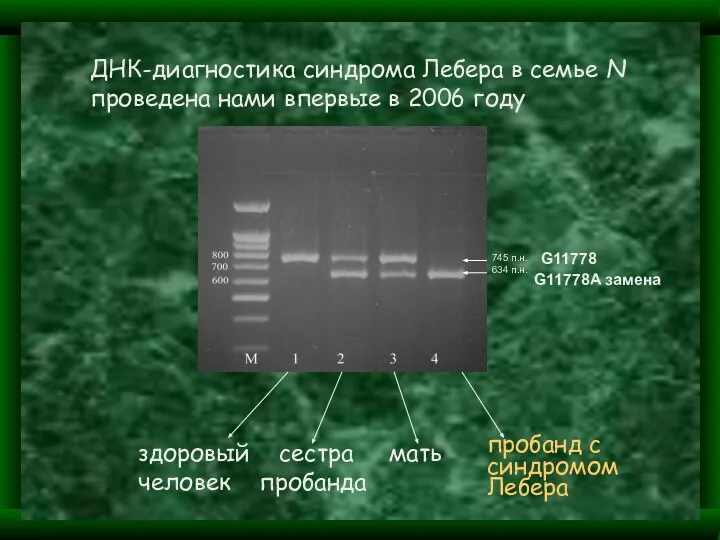
ДНК-диагностика синдрома Лебера в семье N проведена нами впервые в 2006
ДНК-диагностика синдрома Лебера в семье N проведена нами впервые в 2006

Лишь у 50% мужчин и 10% женщин носителей патогенных мутаций комплекса
Лишь у 50% мужчин и 10% женщин носителей патогенных мутаций комплекса

Мутации генов транспортной РНК
Самая часто встречающаяся точечная мутация: А3243G в лейциновой
Мутации генов транспортной РНК
Самая часто встречающаяся точечная мутация: А3243G в лейциновой

Впервые ДНК-диагностика синдрома MELAS была проведена нами в 2007 году
Мама: фенотипически
Впервые ДНК-диагностика синдрома MELAS была проведена нами в 2007 году
Мама: фенотипически

Мутация резко снижает эффективность трансляции в мт и тем самым провоцирует
Мутация резко снижает эффективность трансляции в мт и тем самым провоцирует

Мутации генов рибосомальной РНК
Чаще всего встречается мутация гена 12S
Мутации генов рибосомальной РНК
Чаще всего встречается мутация гена 12S

T8993G: лейцин замещается на аргинин в ATPase6,
что приводит к
T8993G: лейцин замещается на аргинин в ATPase6,
что приводит к

Синдром Лея – тяжелейшее нейродегенеративное заболевание:
- симметричные некротические повреждения в субкортикальных
Синдром Лея – тяжелейшее нейродегенеративное заболевание:
- симметричные некротические повреждения в субкортикальных
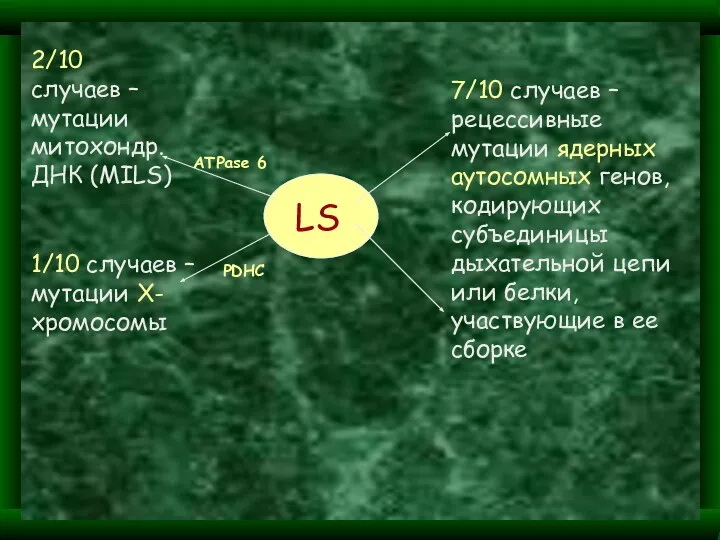
LS
2/10 случаев – мутации митохондр. ДНК (MILS)
1/10 cлучаев – мутации Х-хромосомы
ATPase
LS
2/10 случаев – мутации митохондр. ДНК (MILS)
1/10 cлучаев – мутации Х-хромосомы
ATPase
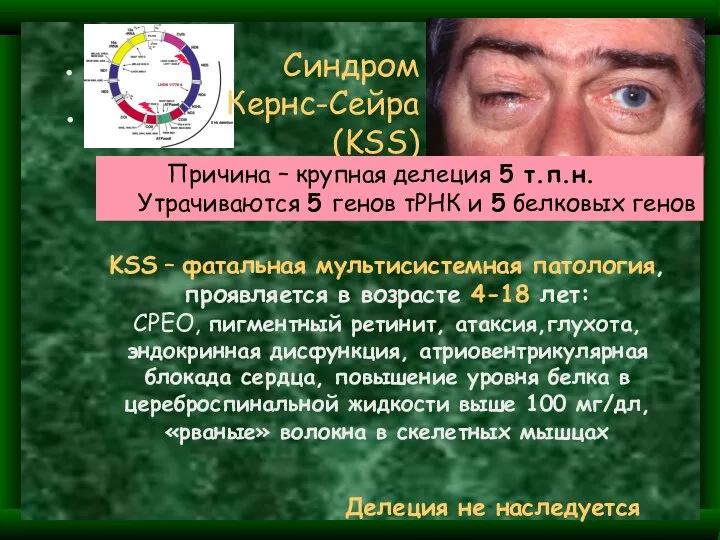

Та же делеция 5 т.п.н. вызывает еще 2 синдрома:
Синдром PEO –
Та же делеция 5 т.п.н. вызывает еще 2 синдрома:
Синдром PEO –
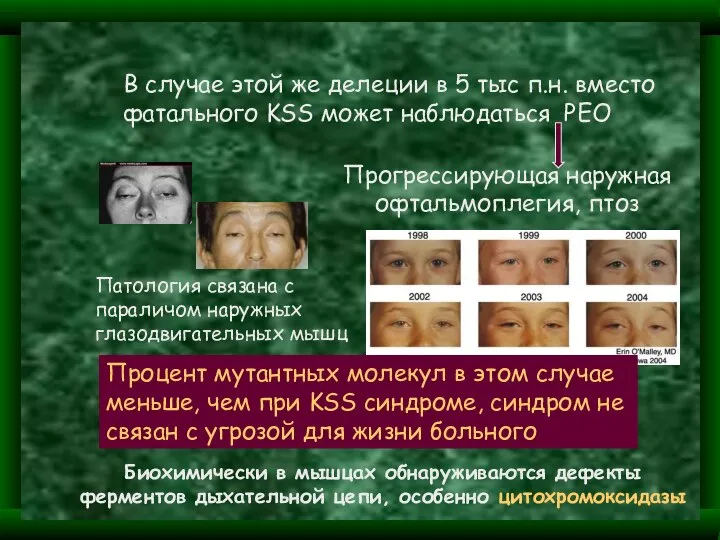
В случае этой же делеции в 5 тыс п.н. вместо фатального
В случае этой же делеции в 5 тыс п.н. вместо фатального

Синдром митохондриальной деплеции - МDS
В клетках остается 1 - 30% от
Синдром митохондриальной деплеции - МDS
В клетках остается 1 - 30% от
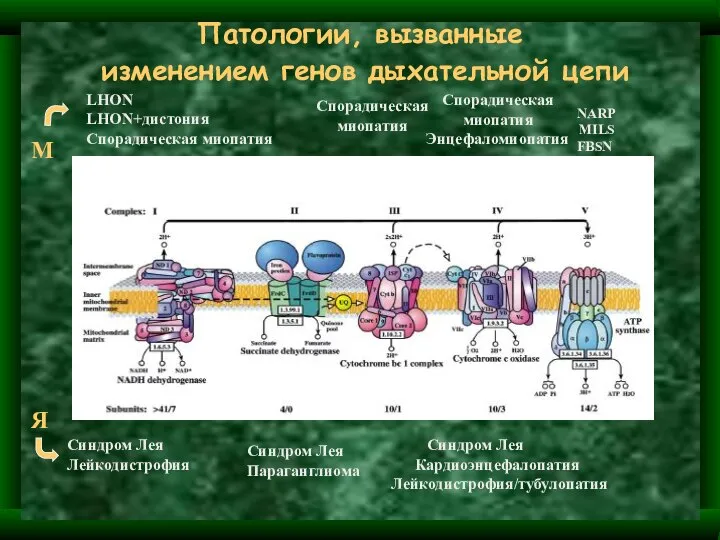
Патологии, вызванные
изменением генов дыхательной цепи
LHON
LHON+дистония
Спорадическая миопатия
Спорадическая миопатия
Спорадическая
Патологии, вызванные
изменением генов дыхательной цепи
LHON
LHON+дистония
Спорадическая миопатия
Спорадическая миопатия
Спорадическая
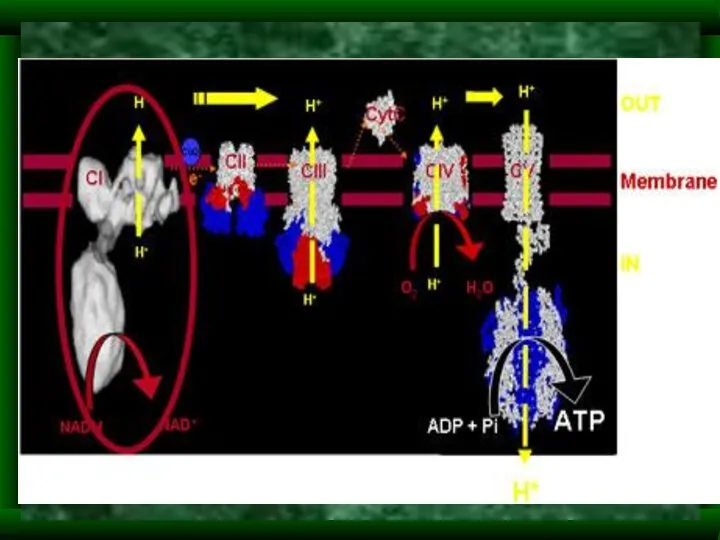



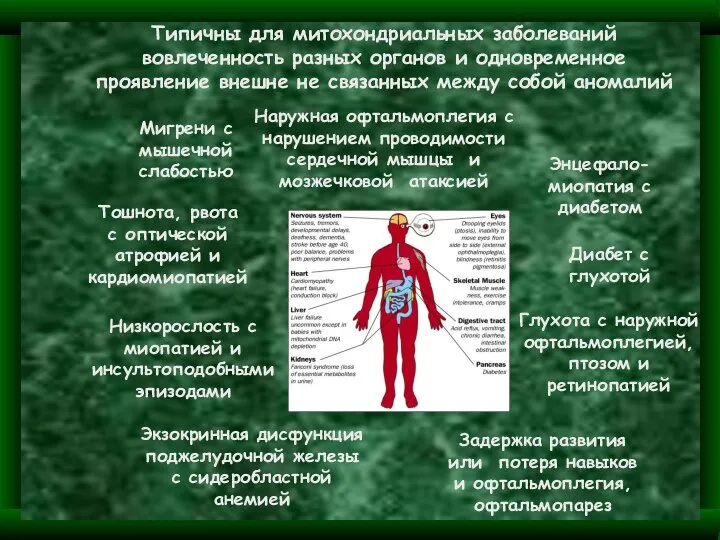




Другой подход - уменьшить соотношение мутантная:нормальная мтДНК
I. Увеличить количество
Другой подход - уменьшить соотношение мутантная:нормальная мтДНК
I. Увеличить количество

II.Уменьшить количество мутантных молекул мтДНК
Разработка синтетических молекул, избирательно связывающихся с мутаными
II.Уменьшить количество мутантных молекул мтДНК
Разработка синтетических молекул, избирательно связывающихся с мутаными

Лечение митохондриальных болезней (4)
Импорт из цитоплазмы нормальных тРНК вместо дефектных митохондриальных
Замена
Лечение митохондриальных болезней (4)
Импорт из цитоплазмы нормальных тРНК вместо дефектных митохондриальных
Замена

Лечение митохондриальных болезней –насколько это реально?
Фармакологическое
Вылечить от митохондриального заболевания сегодня невозможно
Применяется
Лечение митохондриальных болезней –насколько это реально?
Фармакологическое
Вылечить от митохондриального заболевания сегодня невозможно
Применяется

Ряд препаратов провоцирует митохондриальные заболевания или отягощает их течение
Вальпроат:
Ряд препаратов провоцирует митохондриальные заболевания или отягощает их течение
Вальпроат:
 Обеспечение организма кислородом. Выведение углекислого газа и воды
Обеспечение организма кислородом. Выведение углекислого газа и воды Антикоагулянты
Антикоагулянты Проблемы женщин, подвергающихся домашнему насилию
Проблемы женщин, подвергающихся домашнему насилию Микробиоценозы при кишечных инфекциях собак на территории Прибайкалья
Микробиоценозы при кишечных инфекциях собак на территории Прибайкалья Методы исследования слуха
Методы исследования слуха Хронический пиелонефрит. Клиника. Лечение. Участие медсестры в проведении профилактики заболевания
Хронический пиелонефрит. Клиника. Лечение. Участие медсестры в проведении профилактики заболевания Взрывные травмы (общая контузия и контузия головного мозга)
Взрывные травмы (общая контузия и контузия головного мозга) Травмы живота
Травмы живота Бактеріальні інфекції у новонароджених
Бактеріальні інфекції у новонароджених Классификация видов одаренности
Классификация видов одаренности Кишечный шов
Кишечный шов МРТ-диагностика заболеваний позвоночника
МРТ-диагностика заболеваний позвоночника Сестринская помощь при гепатитах Ямбикова
Сестринская помощь при гепатитах Ямбикова Болезни пищеварительной системы
Болезни пищеварительной системы Внутренняя картина болезни
Внутренняя картина болезни Машина құрылысы өнеркәсібіндегі еңбек гигиенасы. Химиялық өнеркәсіптегі еңбек гигиенасы. Өндірістік травматизм
Машина құрылысы өнеркәсібіндегі еңбек гигиенасы. Химиялық өнеркәсіптегі еңбек гигиенасы. Өндірістік травматизм Микрофлора влагалища
Микрофлора влагалища Cardiac emergencies and cpr
Cardiac emergencies and cpr Рожа. Определение. Возбудитель. Классификация
Рожа. Определение. Возбудитель. Классификация Общая и частная психопатология:: определение, методические подходы диагностики, лечения, профилактики
Общая и частная психопатология:: определение, методические подходы диагностики, лечения, профилактики Лекция №5. Методология создания профилактических программ
Лекция №5. Методология создания профилактических программ Эпидемиология и профилактика антропонозов с контактным механизмом передачи
Эпидемиология и профилактика антропонозов с контактным механизмом передачи Инструментальные методы исследования в гинекологии
Инструментальные методы исследования в гинекологии Запам’ятовування, відтворення, забування
Запам’ятовування, відтворення, забування ЛФК в гериатрии
ЛФК в гериатрии Методы диагностики беременности
Методы диагностики беременности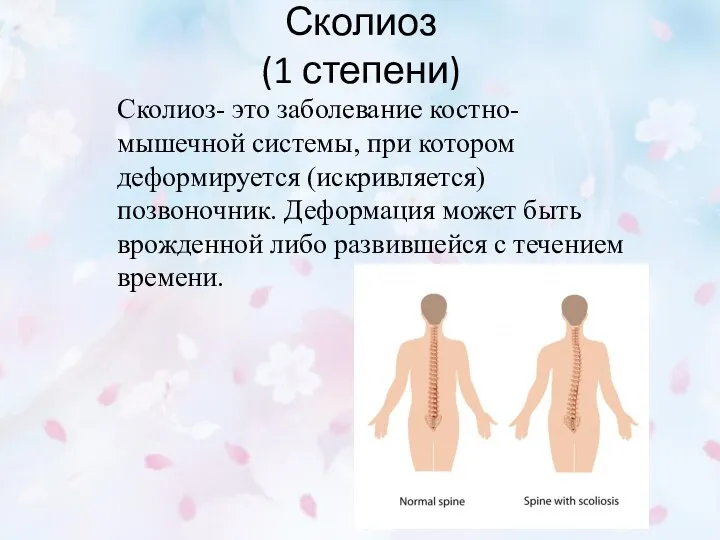 Сколиоз (1 степени)
Сколиоз (1 степени)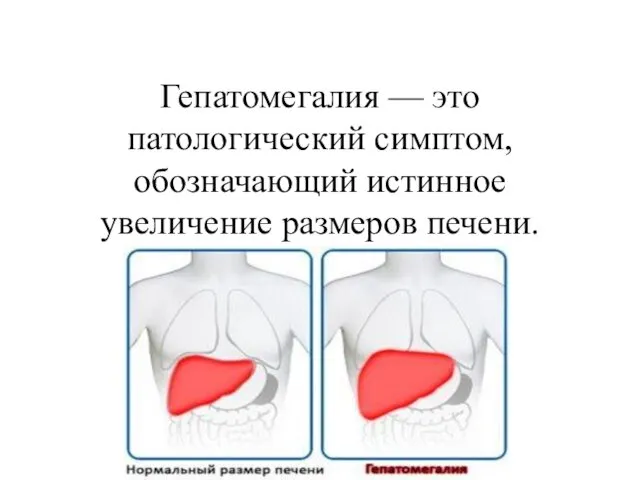 Гепатомегалия: классификация, диагностика, лечение
Гепатомегалия: классификация, диагностика, лечение