- Наследственная патология

Содержание
- 2. О ГЕНОМЕ 20-25 тыс. генов благодаря альтернативному сплайсингу обеспечивают образование более 100 тыс. белков Генетика изучает
- 3. ГЕНЫ И БОЛЕЗНИ ЧЕЛОВЕКА 670 генетических заболеваний приходится на 1000 человек Каждый человек – носитель 5-8
- 6. ПРИЧИНЫ УВЕЛИЧЕНИЯ ЧАСТОТЫ НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИЙ. 1. Ликвидация и уменьшение частоты ряда инфекционные и алиментарных заболеваний.
- 7. ОСНОВНАЯ ПРИЧИНА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ - МУТАЦИЯ. Мутация - внезапное скачкообразное стойкое изменение наследственности. Мутагены: 1. Физические:
- 8. МУТАЦИИ Мутация – стойкое изменение в ДНК. Мутации бывают Генные (изменение структуры ДНК) Хромосомные (изменение структуры
- 9. ГЕННЫЕ МУТАЦИИ Точечные мутации в кодирующих последовательностях I. Замена одной АК на другую Изменение значения последовательности
- 10. ГЕННЫЕ МУТАЦИИ II. Превращение кодирующей АК в стоп-кодон Преждевременная остановка трансляции гена нонсенс-мутация (CAG -> UAG
- 11. ГЕННЫЕ МУТАЦИИ Мутации в некодирующих последовательностях (некоторые формы анемии) – не затрагивают экзоны, но возникают в
- 12. ГЕННЫЕ МУТАЦИИ
- 13. ГЕННЫЕ МУТАЦИИ Делеции Вставки (инсерции) Кроме того, выделяют тринуклеотидные повторы (пример: многочисленные повторы CGG в гене
- 14. ХРОМОСОМНЫЕ МУТАЦИИ
- 15. ГЕНОМНЫЕ МУТАЦИИ
- 16. МУТАЦИИ – ТОЛЬКО ВРЕД? CCR5 – белок, кодируемый одноименным геном на коротком плече 3 хромосомы. Находится
- 18. НАСЛЕДСТВЕННОЕ, СЕМЕЙНОЕ ИЛИ ВРОЖДЁННОЕ ЗАБОЛЕВАНИЕ? Наследственные нарушения происходят от одного из родителей, передаются через гаметы от
- 19. МОНОГЕННОСТЬ И ПОЛИГЕННОСТЬ Моногенные заболевания наследуются по законам Менделя. Типы наследования: аутосомно-доминантный, аутосомно-рецессивный, сцепленный с X-хромосомой.
- 20. АУТОСОМНО-РЕЦЕССИВНЫЕ ЗАБОЛЕВАНИЯ
- 21. ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ Больной мужчина не передает заболевания сыновьям. 50% сыновей гетерозиготной женщины имеют шанс
- 22. ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ
- 25. ОСНОВЫ МОНОГЕННЫХ ЗАБОЛЕВАНИЙ Мутация в гене Аномальный белок или его отсутствие II. Дефекты ферментов (альбинизм, галактоземия,
- 26. СЕМЕЙНАЯ ГИПЕРХОЛЕСТЕРИНЕМИЯ Метаболизм ЛПНП Мутация в гене, кодирующем рецептор к ЛПНП (описано более 900 мутаций в
- 27. РЕГУЛЯЦИЯ МЕТАБОЛИЗМА ХОЛЕСТЕРИНА
- 28. ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ Кислые гидролазы (ферменты лизосом) перерабатывают внутриклеточные органеллы (аутофагия) или внеклеточные субстраты (гетерофагия). При
- 29. ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ II. Дефекты ферментов
- 30. БОЛЕЗНЬ ТЕЯ-САКСА Ганглиозидоз, обусловленный дефицитом фермента гексоаминидазы А. Заболевание наиболее распространено среди евреев, частота 1:30. Лизосомные
- 31. БОЛЕЗНЬ ТЕЯ-САКСА Вишнево-красное пятно на макуле – симптом многих болезней накопления, поражающих нервную систему Лизосомные болезни
- 32. БОЛЕЗНЬ НИМАНА-ПИКА Типы А и В – нет сфингомиелиназы -> накопление сфингомиелина – обязательного компонента клеточных
- 33. БОЛЕЗНЬ НИМАНА-ПИКА Зеброподобные тельца – Переполненные лизосомы Лизосомные болезни накопления Пенистый вид цитоплазмы гепатоцитов – следствие
- 34. БОЛЕЗНЬ ГОШЕ Накопление глюкоцереброзида (компонент клеточных мембран стареющих клеток) в результате мутации в гене, кодирующем глюкоцереброзидазу.
- 36. ХРОМОСОМНЫЕ БОЛЕЗНИ 6:1000. Геномная или хромосомная мутация
- 37. ЧАЩЕ ВСТРЕЧАЮТСЯ: Трисомии: - 1. По 21-й хромосоме - болезнь Дауна нарушение лицевого черепа и мозга
- 38. СТИГМЫ ДИЗЭМБРИОГЕНЕЗА Анатомическое отклонение от строения органа или части тела, проявляется с новорожденного периода. Это результат
- 39. Долихоцефалия, плоский и широкий нос, низкое расположение ушных раковин Полидактилия Готическое нёбо
- 40. ТРИСОМИЯ ПО 21-Й ХРОМОСОМЕ (СИНДРОМ ДАУНА) 47XX, 47XY Родители здоровы. Причина – нерасхождение хромосомы в мейозе.
- 44. ЗАБОЛЕВАНИЯ, ВЫЗВАННЫЕ ПОРАЖЕНИЕМ ПОЛОВЫХ ХРОМОСОМ Синдром Клайнфельтера – мужской гипогонадизм, 47XXY (90%). 1:660 мальчиков. Синдром Тернера
- 46. МОЛЕКУЛЯРНАЯ ДИАГНОСТИКА ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ
- 47. ПОКАЗАНИЯ К ПРЕНАТАЛЬНОЙ ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ Возраст матери >35 лет Один из родителей является носителем мутантного
- 48. ПОКАЗАНИЯ К ПОСТНАТАЛЬНОЙ ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ Множественные врожденные пороки Не объясненная другими причинами умственная отсталость и/или
- 49. МЕТОДЫ МОЛЕКУЛЯРНОЙ ДИАГНОСТИКИ ГЕНОМНЫХ ИЗМЕНЕНИЙ ПЦР Секвенирование ДНК Расширенный геномный анализ Саузерн-блоттинг Метод флуоресцентной гибридизации in
- 51. Скачать презентацию

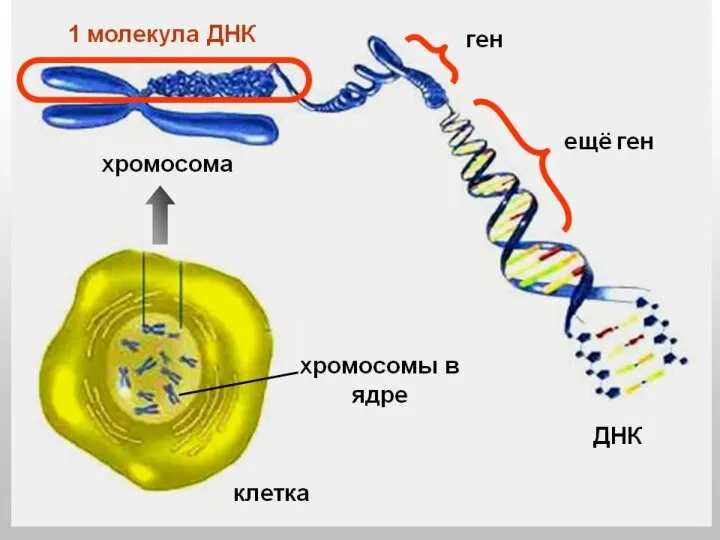
О ГЕНОМЕ
20-25 тыс. генов благодаря альтернативному сплайсингу обеспечивают образование более 100
О ГЕНОМЕ
20-25 тыс. генов благодаря альтернативному сплайсингу обеспечивают образование более 100

ГЕНЫ И БОЛЕЗНИ ЧЕЛОВЕКА
670 генетических заболеваний приходится на 1000 человек
Каждый человек
ГЕНЫ И БОЛЕЗНИ ЧЕЛОВЕКА
670 генетических заболеваний приходится на 1000 человек
Каждый человек


ПРИЧИНЫ УВЕЛИЧЕНИЯ ЧАСТОТЫ НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИЙ.
1. Ликвидация и уменьшение частоты
ПРИЧИНЫ УВЕЛИЧЕНИЯ ЧАСТОТЫ НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИЙ.
1. Ликвидация и уменьшение частоты

ОСНОВНАЯ ПРИЧИНА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ - МУТАЦИЯ.
Мутация - внезапное скачкообразное стойкое
ОСНОВНАЯ ПРИЧИНА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ - МУТАЦИЯ.
Мутация - внезапное скачкообразное стойкое

МУТАЦИИ
Мутация – стойкое изменение в ДНК.
Мутации бывают
Генные (изменение структуры ДНК)
Хромосомные
МУТАЦИИ
Мутация – стойкое изменение в ДНК.
Мутации бывают
Генные (изменение структуры ДНК)
Хромосомные

ГЕННЫЕ МУТАЦИИ
Точечные мутации в кодирующих последовательностях
I. Замена одной АК
ГЕННЫЕ МУТАЦИИ
Точечные мутации в кодирующих последовательностях
I. Замена одной АК

ГЕННЫЕ МУТАЦИИ
II. Превращение кодирующей АК в стоп-кодон
Преждевременная остановка трансляции гена
нонсенс-мутация
(CAG ->
ГЕННЫЕ МУТАЦИИ
II. Превращение кодирующей АК в стоп-кодон
Преждевременная остановка трансляции гена
нонсенс-мутация
(CAG ->

ГЕННЫЕ МУТАЦИИ
Мутации в некодирующих последовательностях
(некоторые формы анемии) – не затрагивают экзоны,
ГЕННЫЕ МУТАЦИИ
Мутации в некодирующих последовательностях
(некоторые формы анемии) – не затрагивают экзоны,
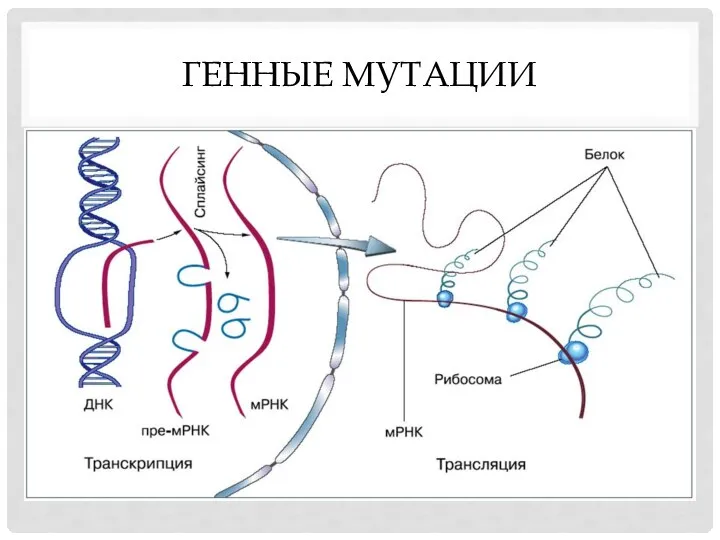
ГЕННЫЕ МУТАЦИИ
ГЕННЫЕ МУТАЦИИ

ГЕННЫЕ МУТАЦИИ
Делеции
Вставки (инсерции)
Кроме того, выделяют тринуклеотидные повторы (пример: многочисленные повторы
ГЕННЫЕ МУТАЦИИ
Делеции
Вставки (инсерции)
Кроме того, выделяют тринуклеотидные повторы (пример: многочисленные повторы
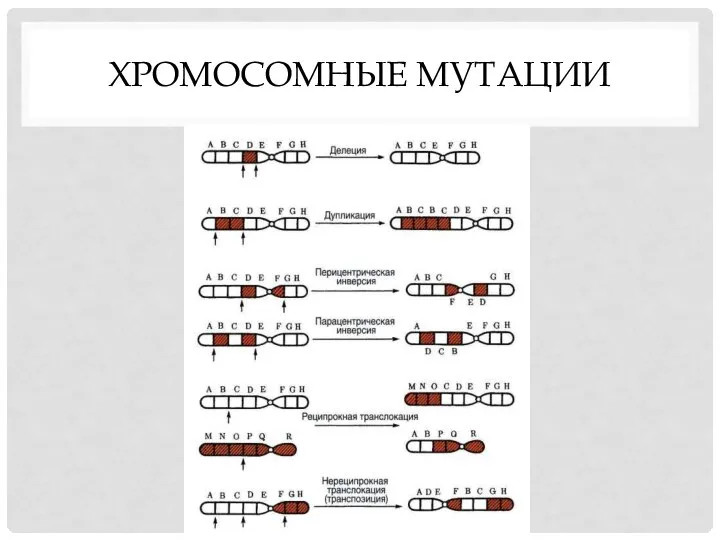
ХРОМОСОМНЫЕ МУТАЦИИ
ХРОМОСОМНЫЕ МУТАЦИИ

ГЕНОМНЫЕ МУТАЦИИ
ГЕНОМНЫЕ МУТАЦИИ

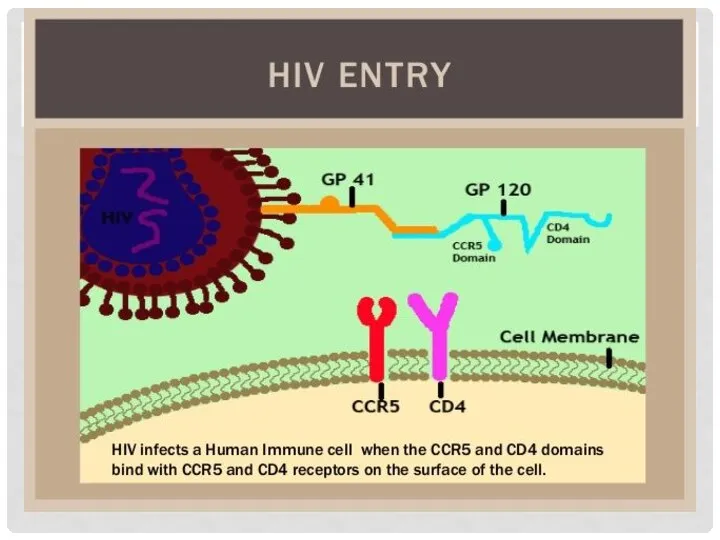
МУТАЦИИ – ТОЛЬКО ВРЕД?
CCR5 – белок, кодируемый одноименным геном на коротком
МУТАЦИИ – ТОЛЬКО ВРЕД?
CCR5 – белок, кодируемый одноименным геном на коротком

НАСЛЕДСТВЕННОЕ, СЕМЕЙНОЕ ИЛИ ВРОЖДЁННОЕ ЗАБОЛЕВАНИЕ?
Наследственные нарушения происходят от одного из родителей,
НАСЛЕДСТВЕННОЕ, СЕМЕЙНОЕ ИЛИ ВРОЖДЁННОЕ ЗАБОЛЕВАНИЕ?
Наследственные нарушения происходят от одного из родителей,

МОНОГЕННОСТЬ И ПОЛИГЕННОСТЬ
Моногенные заболевания наследуются по законам Менделя. Типы наследования: аутосомно-доминантный,
МОНОГЕННОСТЬ И ПОЛИГЕННОСТЬ
Моногенные заболевания наследуются по законам Менделя. Типы наследования: аутосомно-доминантный,

АУТОСОМНО-РЕЦЕССИВНЫЕ ЗАБОЛЕВАНИЯ
АУТОСОМНО-РЕЦЕССИВНЫЕ ЗАБОЛЕВАНИЯ

ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ
Больной мужчина не передает заболевания сыновьям. 50% сыновей
ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ
Больной мужчина не передает заболевания сыновьям. 50% сыновей

ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ
ЗАБОЛЕВАНИЯ, СЦЕПЛЕННЫЕ С Х-ХРОМОСОМОЙ


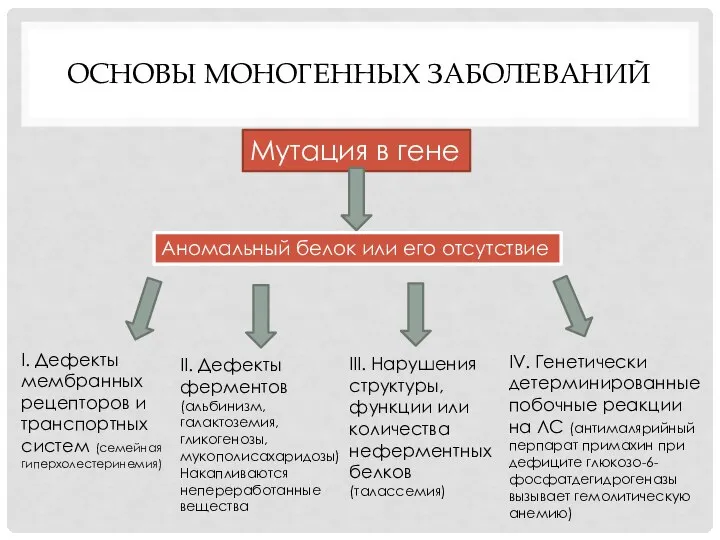
ОСНОВЫ МОНОГЕННЫХ ЗАБОЛЕВАНИЙ
Мутация в гене
Аномальный белок или его отсутствие
II. Дефекты
ферментов
(альбинизм,
ОСНОВЫ МОНОГЕННЫХ ЗАБОЛЕВАНИЙ
Мутация в гене
Аномальный белок или его отсутствие
II. Дефекты
ферментов
(альбинизм,
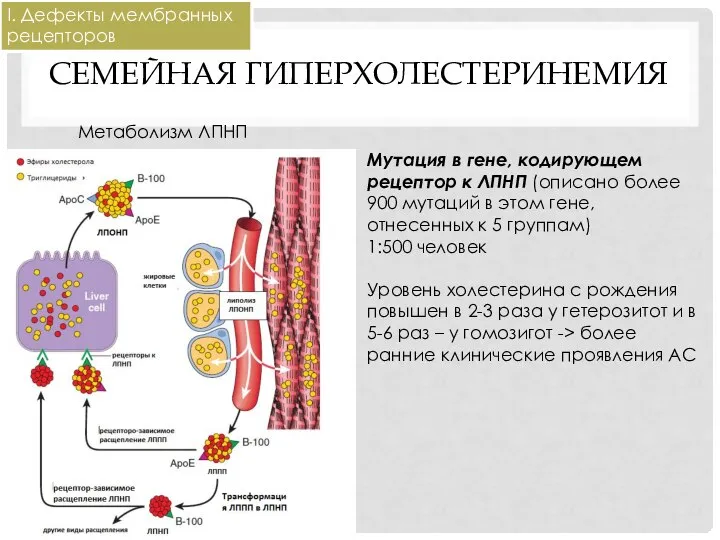
СЕМЕЙНАЯ ГИПЕРХОЛЕСТЕРИНЕМИЯ
Метаболизм ЛПНП
Мутация в гене, кодирующем рецептор к ЛПНП (описано более
СЕМЕЙНАЯ ГИПЕРХОЛЕСТЕРИНЕМИЯ
Метаболизм ЛПНП
Мутация в гене, кодирующем рецептор к ЛПНП (описано более
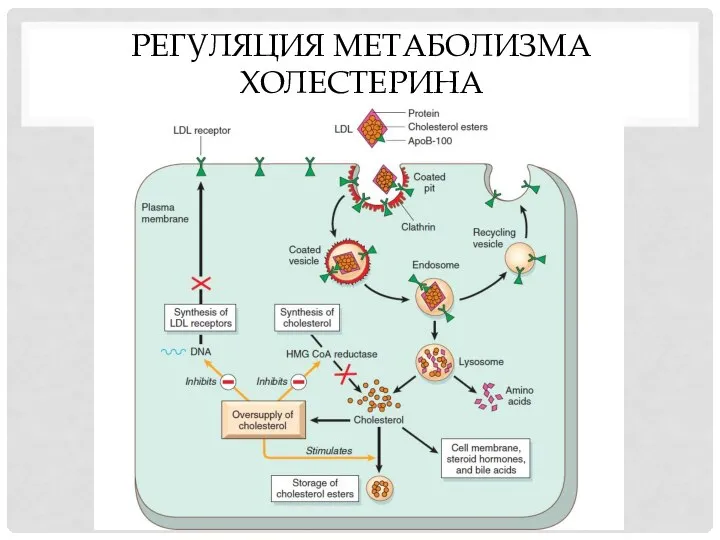
РЕГУЛЯЦИЯ МЕТАБОЛИЗМА ХОЛЕСТЕРИНА
РЕГУЛЯЦИЯ МЕТАБОЛИЗМА ХОЛЕСТЕРИНА

ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ
Кислые гидролазы (ферменты лизосом) перерабатывают внутриклеточные органеллы (аутофагия) или
ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ
Кислые гидролазы (ферменты лизосом) перерабатывают внутриклеточные органеллы (аутофагия) или
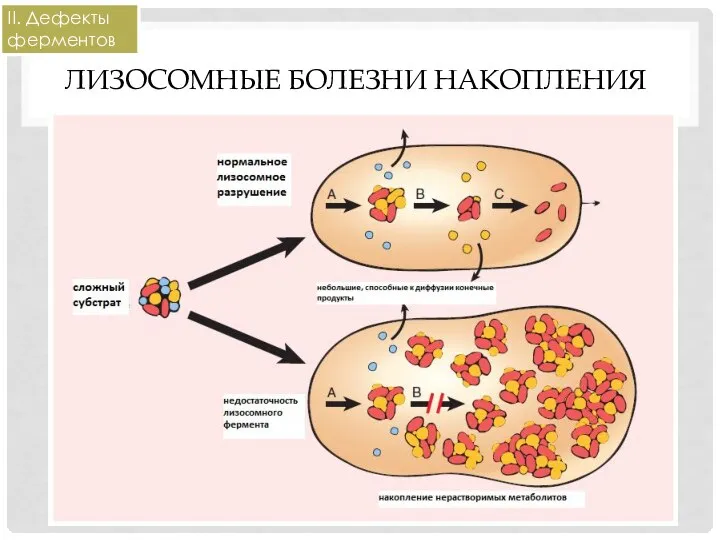
ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ
II. Дефекты ферментов
ЛИЗОСОМНЫЕ БОЛЕЗНИ НАКОПЛЕНИЯ
II. Дефекты ферментов

БОЛЕЗНЬ ТЕЯ-САКСА
Ганглиозидоз, обусловленный дефицитом фермента гексоаминидазы А.
Заболевание наиболее распространено среди
БОЛЕЗНЬ ТЕЯ-САКСА
Ганглиозидоз, обусловленный дефицитом фермента гексоаминидазы А.
Заболевание наиболее распространено среди
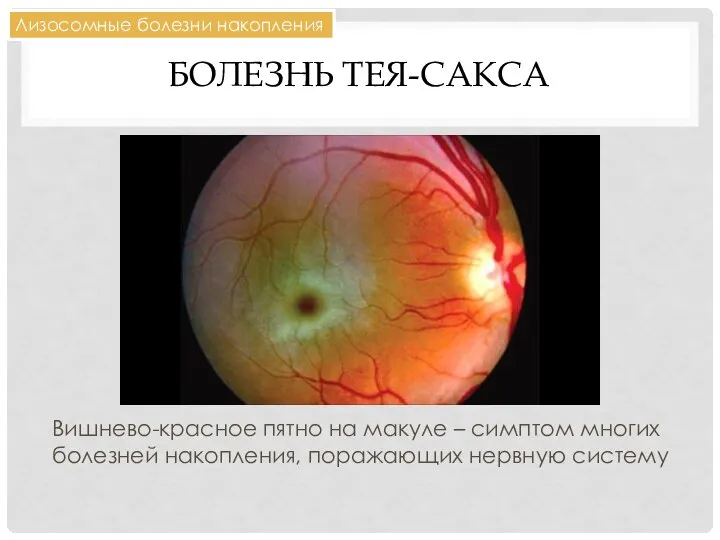
БОЛЕЗНЬ ТЕЯ-САКСА
Вишнево-красное пятно на макуле – симптом многих болезней накопления, поражающих
БОЛЕЗНЬ ТЕЯ-САКСА
Вишнево-красное пятно на макуле – симптом многих болезней накопления, поражающих

БОЛЕЗНЬ НИМАНА-ПИКА
Типы А и В – нет сфингомиелиназы -> накопление сфингомиелина
БОЛЕЗНЬ НИМАНА-ПИКА
Типы А и В – нет сфингомиелиназы -> накопление сфингомиелина
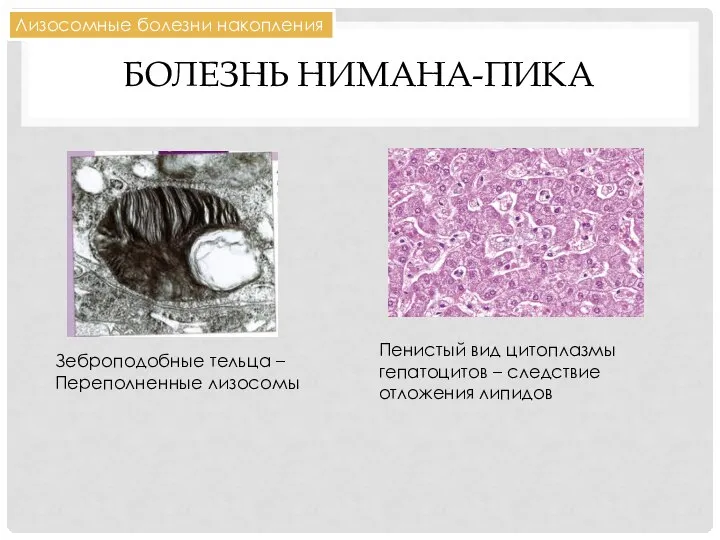
БОЛЕЗНЬ НИМАНА-ПИКА
Зеброподобные тельца –
Переполненные лизосомы
Лизосомные болезни накопления
Пенистый вид цитоплазмы гепатоцитов
БОЛЕЗНЬ НИМАНА-ПИКА
Зеброподобные тельца –
Переполненные лизосомы
Лизосомные болезни накопления
Пенистый вид цитоплазмы гепатоцитов

БОЛЕЗНЬ ГОШЕ
Накопление глюкоцереброзида (компонент клеточных мембран стареющих клеток) в результате мутации
БОЛЕЗНЬ ГОШЕ
Накопление глюкоцереброзида (компонент клеточных мембран стареющих клеток) в результате мутации

ХРОМОСОМНЫЕ БОЛЕЗНИ
6:1000. Геномная или хромосомная мутация
ХРОМОСОМНЫЕ БОЛЕЗНИ
6:1000. Геномная или хромосомная мутация

ЧАЩЕ ВСТРЕЧАЮТСЯ:
Трисомии:
- 1. По 21-й хромосоме - болезнь Дауна
нарушение лицевого
ЧАЩЕ ВСТРЕЧАЮТСЯ:
Трисомии: - 1. По 21-й хромосоме - болезнь Дауна нарушение лицевого

СТИГМЫ ДИЗЭМБРИОГЕНЕЗА
Анатомическое отклонение от строения органа или части тела, проявляется с
СТИГМЫ ДИЗЭМБРИОГЕНЕЗА
Анатомическое отклонение от строения органа или части тела, проявляется с
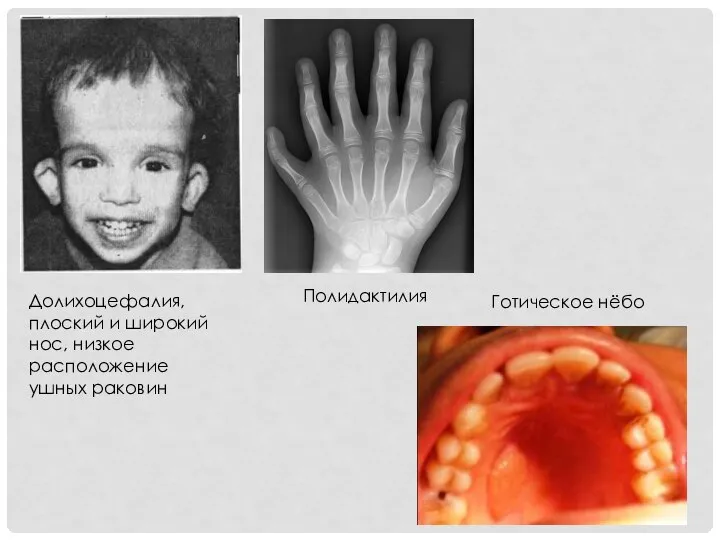
Долихоцефалия, плоский и широкий нос, низкое расположение ушных раковин
Полидактилия
Готическое нёбо
Долихоцефалия, плоский и широкий нос, низкое расположение ушных раковин
Полидактилия
Готическое нёбо

ТРИСОМИЯ ПО 21-Й ХРОМОСОМЕ (СИНДРОМ ДАУНА) 47XX, 47XY
Родители здоровы. Причина –
ТРИСОМИЯ ПО 21-Й ХРОМОСОМЕ (СИНДРОМ ДАУНА) 47XX, 47XY
Родители здоровы. Причина –

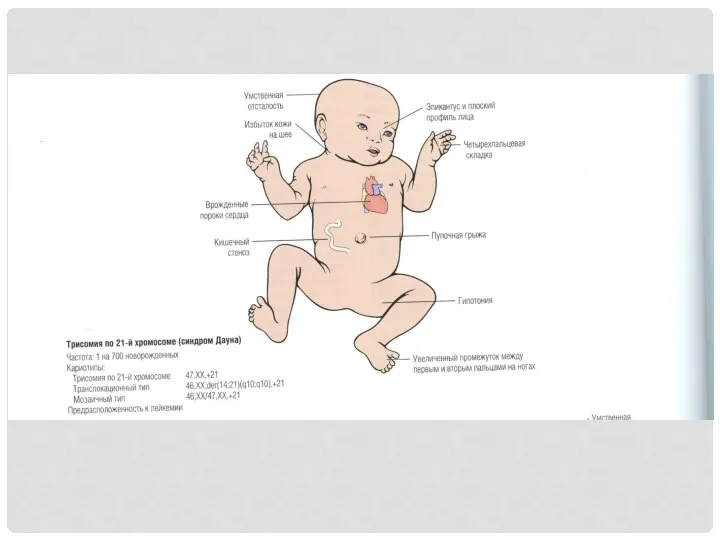
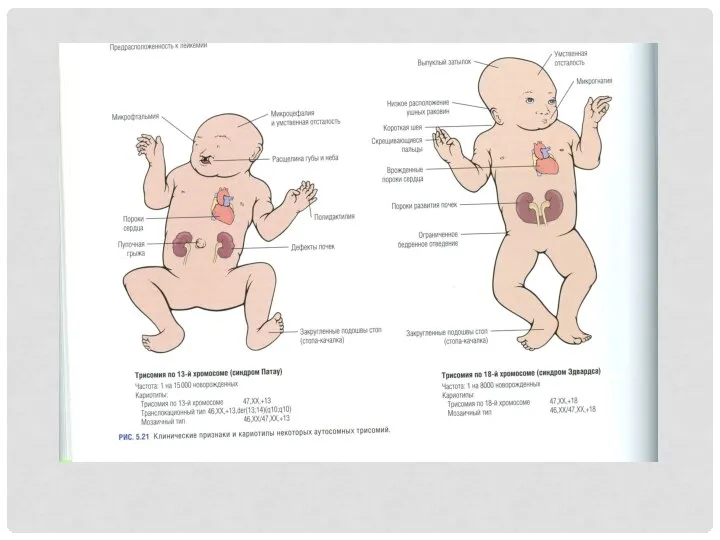

ЗАБОЛЕВАНИЯ, ВЫЗВАННЫЕ ПОРАЖЕНИЕМ ПОЛОВЫХ ХРОМОСОМ
Синдром Клайнфельтера – мужской гипогонадизм, 47XXY (90%).
ЗАБОЛЕВАНИЯ, ВЫЗВАННЫЕ ПОРАЖЕНИЕМ ПОЛОВЫХ ХРОМОСОМ
Синдром Клайнфельтера – мужской гипогонадизм, 47XXY (90%).


МОЛЕКУЛЯРНАЯ ДИАГНОСТИКА ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ
МОЛЕКУЛЯРНАЯ ДИАГНОСТИКА ГЕНЕТИЧЕСКИХ ЗАБОЛЕВАНИЙ

ПОКАЗАНИЯ К ПРЕНАТАЛЬНОЙ ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Возраст матери >35 лет
Один из родителей
ПОКАЗАНИЯ К ПРЕНАТАЛЬНОЙ ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Возраст матери >35 лет
Один из родителей

ПОКАЗАНИЯ К ПОСТНАТАЛЬНОЙ ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Множественные врожденные пороки
Не объясненная другими причинами
ПОКАЗАНИЯ К ПОСТНАТАЛЬНОЙ ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
Множественные врожденные пороки
Не объясненная другими причинами

МЕТОДЫ МОЛЕКУЛЯРНОЙ ДИАГНОСТИКИ ГЕНОМНЫХ ИЗМЕНЕНИЙ
ПЦР
Секвенирование ДНК
Расширенный геномный анализ
Саузерн-блоттинг
Метод флуоресцентной гибридизации in
МЕТОДЫ МОЛЕКУЛЯРНОЙ ДИАГНОСТИКИ ГЕНОМНЫХ ИЗМЕНЕНИЙ
ПЦР
Секвенирование ДНК
Расширенный геномный анализ
Саузерн-блоттинг
Метод флуоресцентной гибридизации in
 Особливості системи виділення в людей різних вікових періодів (Лекція № 8)
Особливості системи виділення в людей різних вікових періодів (Лекція № 8) Средства, влияющие на функцию органов дыхания
Средства, влияющие на функцию органов дыхания Стоматолог тәжірибесінде витаминді препараттар
Стоматолог тәжірибесінде витаминді препараттар Оказание доврачебной помощи
Оказание доврачебной помощи Принципы системы оздоровления
Принципы системы оздоровления Оказание помощи терпящим бедствие на воде
Оказание помощи терпящим бедствие на воде Сахарный диабет и беременность
Сахарный диабет и беременность Менингококковая инфекция
Менингококковая инфекция Features of childrens` blood. The semiotics of major hematological syndromes
Features of childrens` blood. The semiotics of major hematological syndromes Platelet granules
Platelet granules Сүт безі қатерлі ісігі
Сүт безі қатерлі ісігі Аускультация тәсілінің даму тарихы
Аускультация тәсілінің даму тарихы Агрессия у детей и подростков
Агрессия у детей и подростков Коронавирус. Профилактика коронавирусной инфекции
Коронавирус. Профилактика коронавирусной инфекции Вегетарианство
Вегетарианство Активное слушание
Активное слушание Нарушения функции почек
Нарушения функции почек Изучение ощущения счастья беременных женщин в браке и вне брака
Изучение ощущения счастья беременных женщин в браке и вне брака Типовые нарушения функций почек
Типовые нарушения функций почек Электромагниттік сәулеленудің адам ағзасына әсері
Электромагниттік сәулеленудің адам ағзасына әсері Медицинские информационные системы
Медицинские информационные системы Профилактика новой коронавирусной инфекции
Профилактика новой коронавирусной инфекции ЭМ - технология. Биосанация жилых помещений и повышение иммунитета
ЭМ - технология. Биосанация жилых помещений и повышение иммунитета Кровотечение. Классификация
Кровотечение. Классификация Сестринский процесс при хроническом гепатите
Сестринский процесс при хроническом гепатите Как и про что разговаривать с новым поколением
Как и про что разговаривать с новым поколением Бауыр жетіспеушілігі
Бауыр жетіспеушілігі Приобретенные митральные пороки сердца
Приобретенные митральные пороки сердца