- The leukemia

Содержание
- 2. TERMINOLOGY A malignant proliferation of monoclonal hematopoetic cells with accumulation of abnormal immature cells which replace
- 3. Leukemias A very heterogeneic group of disorders, can be classified on a basis of clinical course
- 4. AML – FAB classification M0 – undifferentiated M1 – early myeloblastic M2 – myelocytic M3 –
- 5. FAB classification This classification is based mostly on morphology and immunophenotyping of the blasts Has clinical
- 6. Cytogenetics Cytogenetics is the most important prognostic feature of AML “Favorable” – M2 with t(8;21), M3
- 7. AML – WHO classification AML with recurrent cytogenetic translocations – M2 with t(8;21), M3 with t(15;17)
- 8. ALL The FAB classification is not in use Is classified by the phenotype of the blasts
- 9. ETIOLOGY Environment: irradiation, chemotherapeutic agents, organic solvents – benzene etc. Genetic diseases: neurofibromatosis, Wiscott-Aldrich synd., defective
- 10. CLINICAL FEAURES AML – 1.2% of all cancer deaths in US (about 9200 new cases per
- 11. CLINICAL FEATURES The presenting signs are not specific: Anemia – pallor, weakness, dispnoea Neutropenia – fever,
- 12. LABORATORY Leukocytosis with blasts Metabolic and electrolyte derangement hyperuricemia, hyperkalemia, hyperphosphatemia – tumor lysis syndrome Coagulopathy
- 13. DIAGNOSIS Blasts in blood or bone marrow smear, Auer rods pathognomonic to AML Immunohistochemistry – peroxidase
- 14. Immunophenotyping CD – Cluster Designation, molecules on the surface of the cell, characteristic to each cell
- 15. AML - TREATMENT AML – induction with ARA-C and daunorubicin (7:3); consolidations with HIDAC and others,
- 16. ALL - TREATMENT Protocols based on treatment of childhood ALL, prolonged and intensive therapy with CNS
- 17. CML A clonal expansion of hematopoetic progenitors, characterized clinically by myeloid hyperplasia, leukocytosis with basophilia and
- 18. C M L A phasic disease – chronic phase, accelerated phase, blast crisis Incidence – 1-2:100000
- 19. CML - cytogenetics The first malignancy in which the link between a chromosomal abnormality and leukemogenesis
- 21. CML Philadelphia chromosome – a short chromosome 22discovered at 1960 by Novel and Henderford First chromosomal
- 23. CML pathogenesis The normal product of Abl gene is a protein of 145kd with a week
- 24. Clinical features Most patients are asymptomatic at diagnosis Splenomegaly ± symptoms, anemia, hepatomegaly, purpura, constitutional symptoms
- 25. Laboratory Peripheral blood : leukocytosis with “left shift”, basophillia, eeosinophilia, thrombocytosis, anemia Bone marrow: myeloid (M:E>3:1),
- 26. Laboratory LAP (leukocyte alkaline phosphatase)↓ Transcobalamine↑ Uric acid↑ Cytogenetics - Ph+ {t(9;22)} Molecular - bcr/abl +
- 27. Accelerated Phase ↑Leukocytosis under treatment ↑Basophilia (>20% basophils and eosinophils >10% blasts in peripheral blood >20%
- 28. BLAST CRISIS Developes in 75-80% of patients Median time from diagnosis 3-5 years constitutional symptoms, bone
- 29. TREATMENT Tyrosine kinase inhibitors - glyvec (imatinib mesylate), nilotinib, dasatinib etc., major cytogenetic and molecular responses
- 30. C L L A progressive accumulation of functionally incompetent mature lymphocytes 15-20% of all leukemias, M:F=1.7:1
- 31. C L L Frequent family history of CLL, other B-cell malignancies, autoimmune disorders No other risk
- 32. C L L Immunophenotyping: B-cell markers CD19, CD20, CD21, CD23, ; T-cell marker CD5 is a
- 33. Clinical Manifestations Autoimmune features - Coomb’s+ hemolytic anemia, ITP Recurrent infections - due to hypogammaglobulinemia Symptoms:
- 34. Laboratory Findings >5000 mature appearing lymphocytes Anemia, thrombocytopenia Bone marrow - infiltration by same lymphocytes, decrease
- 35. Diagnostic Criteria Absolute lymphocytosis >5000/ml on few consecutive tests At least 30% lymphocytes in normo- or
- 36. CLL - Staging - Rai System Stage 0 - lymphocytosis blood,marrow Stage 1 - lymphocytosis +
- 37. CLL - Staging Binet Stage A - lymphocytosis and two or less areas of enlarged lymph
- 38. CLL -Treatment Rai st. 0-2 or Binet st. A-B ⇒ observe every 3-6 months, treat if
- 39. Treatment Options Chemotherapy - steroids, alkylating agents ± steroids, purine analogues - fludarabine, combinations Monoclonal antibodies
- 40. CLL - Prognosis Extremely variable - some have progressive course and die within 2-3 years, some
- 41. Richter’s Syndrome In 3-5% the disease undergoes a transformation into aggressive lymphoma - diffuse large cell
- 43. Скачать презентацию

TERMINOLOGY
A malignant proliferation of monoclonal hematopoetic cells with accumulation of abnormal
TERMINOLOGY
A malignant proliferation of monoclonal hematopoetic cells with accumulation of abnormal

Leukemias
A very heterogeneic group of disorders, can be classified on a
Leukemias
A very heterogeneic group of disorders, can be classified on a

AML – FAB classification
M0 – undifferentiated
M1 – early myeloblastic
M2 – myelocytic
M3
AML – FAB classification
M0 – undifferentiated
M1 – early myeloblastic
M2 – myelocytic
M3

FAB classification
This classification is based mostly on morphology and immunophenotyping of
FAB classification
This classification is based mostly on morphology and immunophenotyping of

Cytogenetics
Cytogenetics is the most important prognostic feature of AML
“Favorable” –
Cytogenetics
Cytogenetics is the most important prognostic feature of AML
“Favorable” –

AML – WHO classification
AML with recurrent cytogenetic translocations – M2 with
AML – WHO classification
AML with recurrent cytogenetic translocations – M2 with

ALL
The FAB classification is not in use
Is classified by the phenotype
ALL
The FAB classification is not in use
Is classified by the phenotype

ETIOLOGY
Environment: irradiation, chemotherapeutic agents, organic solvents – benzene etc.
Genetic diseases:
ETIOLOGY
Environment: irradiation, chemotherapeutic agents, organic solvents – benzene etc.
Genetic diseases:

CLINICAL FEAURES
AML – 1.2% of all cancer deaths in US (about
CLINICAL FEAURES
AML – 1.2% of all cancer deaths in US (about

CLINICAL FEATURES
The presenting signs are not specific:
Anemia – pallor, weakness, dispnoea
Neutropenia
CLINICAL FEATURES
The presenting signs are not specific:
Anemia – pallor, weakness, dispnoea
Neutropenia

LABORATORY
Leukocytosis with blasts
Metabolic and electrolyte derangement hyperuricemia, hyperkalemia, hyperphosphatemia – tumor
LABORATORY
Leukocytosis with blasts
Metabolic and electrolyte derangement hyperuricemia, hyperkalemia, hyperphosphatemia – tumor

DIAGNOSIS
Blasts in blood or bone marrow smear, Auer rods pathognomonic to
DIAGNOSIS
Blasts in blood or bone marrow smear, Auer rods pathognomonic to

Immunophenotyping
CD – Cluster Designation, molecules on the surface of the cell,
Immunophenotyping
CD – Cluster Designation, molecules on the surface of the cell,

AML - TREATMENT
AML – induction with ARA-C and daunorubicin (7:3); consolidations
AML - TREATMENT
AML – induction with ARA-C and daunorubicin (7:3); consolidations

ALL - TREATMENT
Protocols based on treatment of childhood ALL, prolonged and
ALL - TREATMENT
Protocols based on treatment of childhood ALL, prolonged and

CML
A clonal expansion of hematopoetic progenitors, characterized clinically by myeloid hyperplasia,
CML
A clonal expansion of hematopoetic progenitors, characterized clinically by myeloid hyperplasia,

C M L
A phasic disease – chronic phase, accelerated phase, blast
C M L
A phasic disease – chronic phase, accelerated phase, blast

CML - cytogenetics
The first malignancy in which the link between a
CML - cytogenetics
The first malignancy in which the link between a

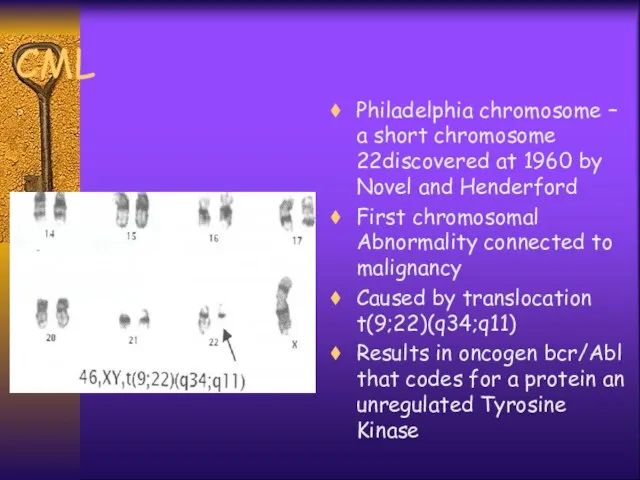
CML
Philadelphia chromosome – a short chromosome 22discovered at 1960 by Novel
CML
Philadelphia chromosome – a short chromosome 22discovered at 1960 by Novel


CML pathogenesis
The normal product of Abl gene is a protein of
CML pathogenesis
The normal product of Abl gene is a protein of

Clinical features
Most patients are asymptomatic at diagnosis
Splenomegaly ± symptoms, anemia, hepatomegaly,
Clinical features
Most patients are asymptomatic at diagnosis
Splenomegaly ± symptoms, anemia, hepatomegaly,
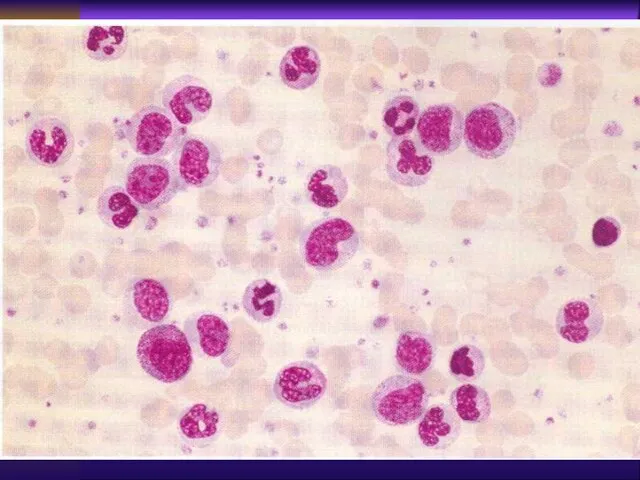
Laboratory
Peripheral blood : leukocytosis with “left shift”, basophillia, eeosinophilia, thrombocytosis,
Laboratory
Peripheral blood : leukocytosis with “left shift”, basophillia, eeosinophilia, thrombocytosis,

Laboratory
LAP (leukocyte alkaline phosphatase)↓
Transcobalamine↑
Uric acid↑
Cytogenetics - Ph+ {t(9;22)}
Molecular - bcr/abl +
Gene
Laboratory
LAP (leukocyte alkaline phosphatase)↓
Transcobalamine↑
Uric acid↑
Cytogenetics - Ph+ {t(9;22)}
Molecular - bcr/abl +
Gene

Accelerated Phase
↑Leukocytosis under treatment
↑Basophilia (>20% basophils and eosinophils
>10% blasts in peripheral
Accelerated Phase
↑Leukocytosis under treatment
↑Basophilia (>20% basophils and eosinophils
>10% blasts in peripheral

BLAST CRISIS
Developes in 75-80% of patients
Median time from diagnosis 3-5 years
constitutional
BLAST CRISIS
Developes in 75-80% of patients
Median time from diagnosis 3-5 years
constitutional

TREATMENT
Tyrosine kinase inhibitors - glyvec (imatinib mesylate), nilotinib, dasatinib etc., major
TREATMENT
Tyrosine kinase inhibitors - glyvec (imatinib mesylate), nilotinib, dasatinib etc., major

C L L
A progressive accumulation of functionally incompetent mature lymphocytes
15-20% of
C L L
A progressive accumulation of functionally incompetent mature lymphocytes
15-20% of

C L L
Frequent family history of CLL, other B-cell malignancies, autoimmune
C L L
Frequent family history of CLL, other B-cell malignancies, autoimmune

C L L
Immunophenotyping: B-cell markers CD19, CD20, CD21, CD23, ; T-cell
C L L
Immunophenotyping: B-cell markers CD19, CD20, CD21, CD23, ; T-cell

Clinical Manifestations
Autoimmune features - Coomb’s+ hemolytic anemia, ITP
Recurrent infections - due
Clinical Manifestations
Autoimmune features - Coomb’s+ hemolytic anemia, ITP
Recurrent infections - due

Laboratory Findings
>5000 mature appearing lymphocytes
Anemia, thrombocytopenia
Bone marrow - infiltration
Laboratory Findings
>5000 mature appearing lymphocytes
Anemia, thrombocytopenia
Bone marrow - infiltration

Diagnostic Criteria
Absolute lymphocytosis >5000/ml on few consecutive tests
At least 30% lymphocytes
Diagnostic Criteria
Absolute lymphocytosis >5000/ml on few consecutive tests
At least 30% lymphocytes
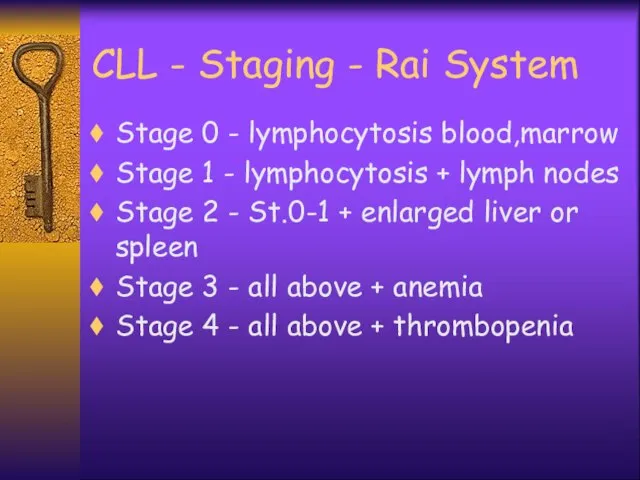
CLL - Staging - Rai System
Stage 0 - lymphocytosis blood,marrow
Stage 1
CLL - Staging - Rai System
Stage 0 - lymphocytosis blood,marrow
Stage 1

CLL - Staging Binet
Stage A - lymphocytosis and two or less
CLL - Staging Binet
Stage A - lymphocytosis and two or less

CLL -Treatment
Rai st. 0-2 or Binet st. A-B ⇒ observe every
CLL -Treatment
Rai st. 0-2 or Binet st. A-B ⇒ observe every

Treatment Options
Chemotherapy - steroids, alkylating agents ± steroids, purine analogues -
Treatment Options
Chemotherapy - steroids, alkylating agents ± steroids, purine analogues -

CLL - Prognosis
Extremely variable - some have progressive course and die
CLL - Prognosis
Extremely variable - some have progressive course and die

Richter’s Syndrome
In 3-5% the disease undergoes a transformation into aggressive lymphoma
Richter’s Syndrome
In 3-5% the disease undergoes a transformation into aggressive lymphoma
 Кожа. Заболевания кожи
Кожа. Заболевания кожи Спирография. Понятие внешнего дыхания
Спирография. Понятие внешнего дыхания Противогрибковые и противовирусные лечебные средства
Противогрибковые и противовирусные лечебные средства Возрастные особенности роста и физического развития подростков (проверочные тесты )
Возрастные особенности роста и физического развития подростков (проверочные тесты ) Опасен ли целлюлит для здоровья
Опасен ли целлюлит для здоровья Симптоматология гепатитов, цирроза печени, холецистита, дискипезии желчевыводящих путей
Симптоматология гепатитов, цирроза печени, холецистита, дискипезии желчевыводящих путей Паразиты
Паразиты Истерия
Истерия Хронический пульпит
Хронический пульпит Влияние излучения, исходящего из сотового телефона на организм человека
Влияние излучения, исходящего из сотового телефона на организм человека Опухоли кроветворной ткани
Опухоли кроветворной ткани Эпидемиология и профилактика антропонозов с контактным механизмом передачи
Эпидемиология и профилактика антропонозов с контактным механизмом передачи Возрастные особенности суицидального поведения
Возрастные особенности суицидального поведения Жасқа дейінгі балардың психо-моторлы даму ерекшеліктері
Жасқа дейінгі балардың психо-моторлы даму ерекшеліктері Периферический и центральный отделы речевого аппарата
Периферический и центральный отделы речевого аппарата Чувства, эмоции
Чувства, эмоции Реанимация и интенсивная терапия острых отравлений. Особенности реанимационного пособия при несчастных случаях
Реанимация и интенсивная терапия острых отравлений. Особенности реанимационного пособия при несчастных случаях Первая помощь пострадавшим и её значение
Первая помощь пострадавшим и её значение Презентация по медицине Термические повреждения,ожоги, электротравмы
Презентация по медицине Термические повреждения,ожоги, электротравмы  Анонимные Наркоманы
Анонимные Наркоманы Выготский Лев Семёнович (1896—1934)
Выготский Лев Семёнович (1896—1934) Острые бактериальные воздушно-капельные инфекции
Острые бактериальные воздушно-капельные инфекции Эпилепсия
Эпилепсия Соматика Томаса Ханны и метод Фельденкрайза
Соматика Томаса Ханны и метод Фельденкрайза Мировые стандарты качества и безопасности продуктов питания
Мировые стандарты качества и безопасности продуктов питания Нейровизуализация в неврологии
Нейровизуализация в неврологии Новое слово в иммунологии! вторая жизнь сенсационного научного открытия прошлого века
Новое слово в иммунологии! вторая жизнь сенсационного научного открытия прошлого века Шаблоны для проведения имплантации зубов
Шаблоны для проведения имплантации зубов