- Мониторинг НС при применении медицинских изделий, заявляемых на регистрацию в ЕАЭС

Содержание
- 2. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий, утвержденные решением Совета Евразийской экономической комиссии
- 3. Соглашение о единых принципах и правилах обращения медицинских изделий в рамках Евразийского экономического союза от 23.12.2014
- 4. ПРАВИЛА проведения мониторинга безопасности, качества и эффективности медицинских изделий, утвержденные решением Коллегии Евразийской экономической комиссии от
- 5. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 3: "безопасность медицинских изделий" -
- 6. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 5: До подачи в уполномоченный
- 7. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 18: Уполномоченным органом (экспертной организацией)
- 8. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий, пункт 18 Экспертная организация уведомляет заявителя
- 9. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 20: Ответственность за достоверность предоставленного
- 10. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 24: Проведение экспертизы медицинского изделия
- 11. ПРАВИЛА проведения мониторинга безопасности, качества и эффективности медицинских изделий
- 12. Цели и задачи пострегистрационного клинического мониторинга Имеющиеся клинические данные Специфические особенности, связанные с медицинским изделием Факторы
- 13. Схема плана пострегистрационного клинического мониторинга Методы (способ) получения клинических данных и их обоснование Методы (способы) статистического
- 14. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 24: Проведение экспертизы медицинского изделия
- 15. Требованиями к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска
- 16. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 24: Проведение экспертизы медицинского изделия
- 17. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий, пункт 25 уполномоченный орган (экспертная организация)
- 18. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 27: Основаниями для вынесения уполномоченным
- 19. План пострегистра-ционного клинического мониторинга Сообщения о НС и ответные корректиру-ющие действия План сбора и анализа данных
- 20. ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 17: Заявитель представляет в уполномоченный
- 21. Сообщения о неблагоприятных событиях и ответных корректирующих действиях
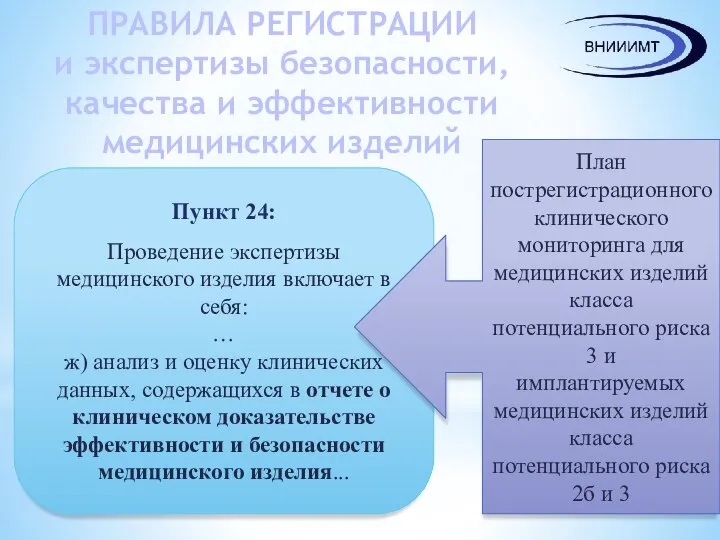
- 22. План постегистрационного клинического мониторинга для медицинских изделий класса потенциального риска 3 и имплантируемых медицинских изделий класса
- 23. План отслеживании и выявлении побочных действий медицинских изделий в процессе их эксплуатации
- 24. «Особые» медицинские изделия Комплекты, наборы МИ, в состав которых входят уже зарегистрированные МИ (аптечка, набор инструментов,
- 25. Источники информации о неблагоприятных событиях ? FDA, США MHRA, Великобритания Health Canada, Канада Swiss medic, Швейцария
- 26. Источники информации о неблагоприятных событиях
- 28. Скачать презентацию

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий,
утвержденные решением Совета
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий,
утвержденные решением Совета

Соглашение о единых принципах и правилах обращения медицинских изделий
в рамках Евразийского
Соглашение о единых принципах и правилах обращения медицинских изделий в рамках Евразийского

ПРАВИЛА
проведения мониторинга безопасности,
качества и эффективности медицинских изделий,
утвержденные решением Коллегии Евразийской экономической
ПРАВИЛА
проведения мониторинга безопасности,
качества и эффективности медицинских изделий,
утвержденные решением Коллегии Евразийской экономической
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности,
качества и эффективности
медицинских изделий
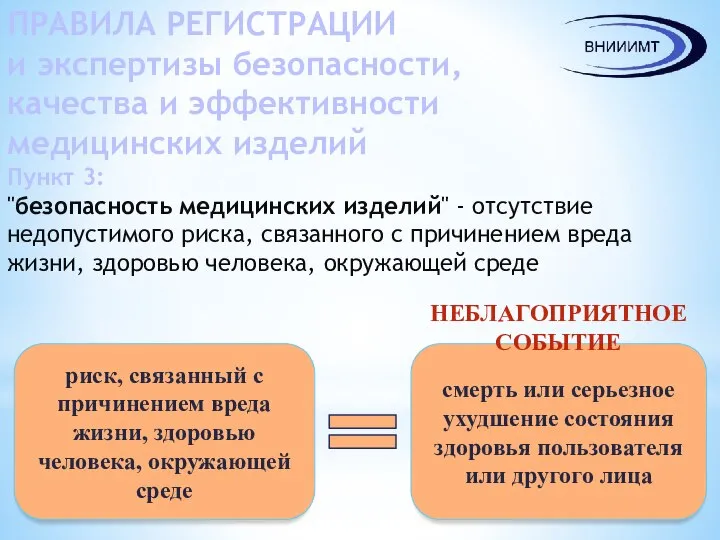
Пункт 3:
"безопасность медицинских изделий"
ПРАВИЛА РЕГИСТРАЦИИ и экспертизы безопасности, качества и эффективности медицинских изделий Пункт 3: "безопасность медицинских изделий"

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 5:
До подачи
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 5:
До подачи

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
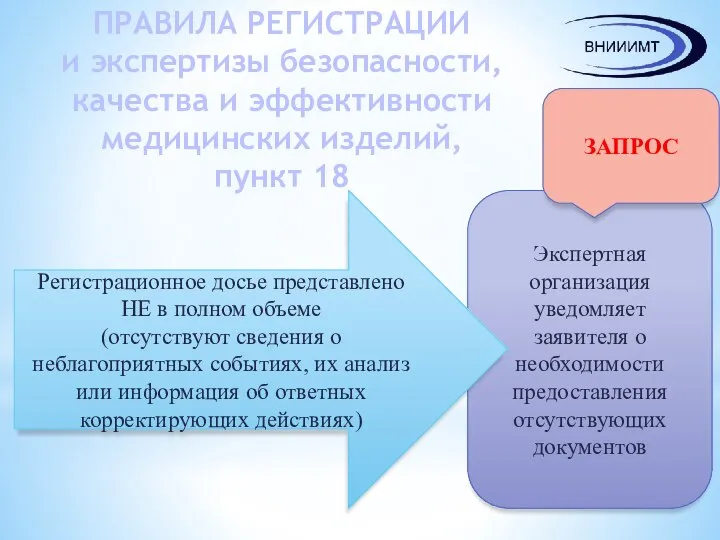
Пункт 18:
Уполномоченным органом
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 18:
Уполномоченным органом
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий,
пункт 18
Экспертная организация
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий,
пункт 18
Экспертная организация

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 20:
Ответственность за
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 20:
Ответственность за
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 24:
Проведение экспертизы
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 24:
Проведение экспертизы
ПРАВИЛА проведения мониторинга безопасности, качества и эффективности медицинских изделий
ПРАВИЛА проведения мониторинга безопасности, качества и эффективности медицинских изделий

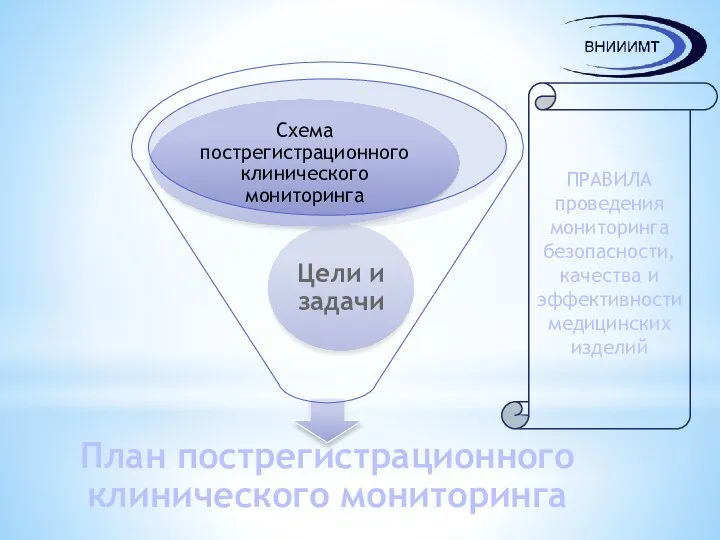
Цели и задачи пострегистрационного клинического мониторинга
Имеющиеся клинические данные
Специфические особенности, связанные с
Цели и задачи пострегистрационного клинического мониторинга
Имеющиеся клинические данные
Специфические особенности, связанные с

Схема плана пострегистрационного клинического мониторинга
Методы (способ) получения клинических данных и их
Схема плана пострегистрационного клинического мониторинга
Методы (способ) получения клинических данных и их

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 24:
Проведение экспертизы
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 24:
Проведение экспертизы

Требованиями к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий
Требованиями к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 24: Проведение
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 24: Проведение
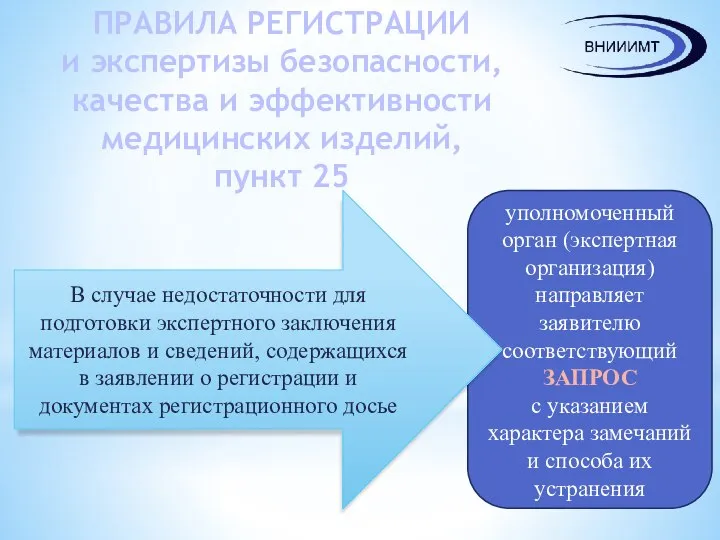
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий,
пункт 25
уполномоченный орган
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий,
пункт 25
уполномоченный орган

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 27:
Основаниями для
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 27:
Основаниями для

План пострегистра-ционного клинического мониторинга
Сообщения о НС и ответные корректиру-ющие действия
План сбора
План пострегистра-ционного клинического мониторинга
Сообщения о НС и ответные корректиру-ющие действия
План сбора

ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 17:
Заявитель представляет
ПРАВИЛА РЕГИСТРАЦИИ
и экспертизы безопасности, качества и эффективности медицинских изделий
Пункт 17:
Заявитель представляет
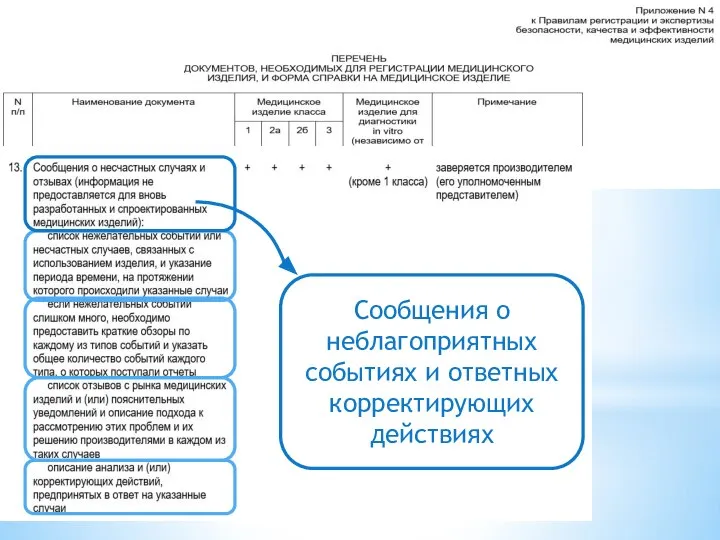
Сообщения о неблагоприятных событиях и ответных корректирующих действиях
Сообщения о неблагоприятных событиях и ответных корректирующих действиях
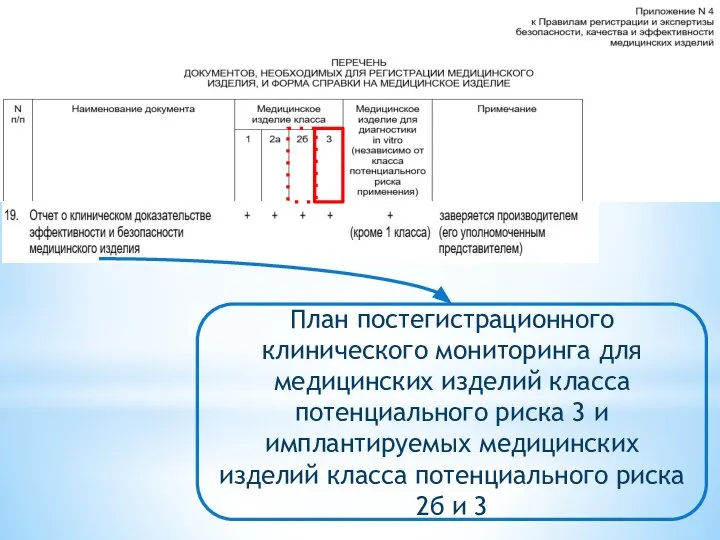
План постегистрационного клинического мониторинга для медицинских изделий класса потенциального риска 3
План постегистрационного клинического мониторинга для медицинских изделий класса потенциального риска 3
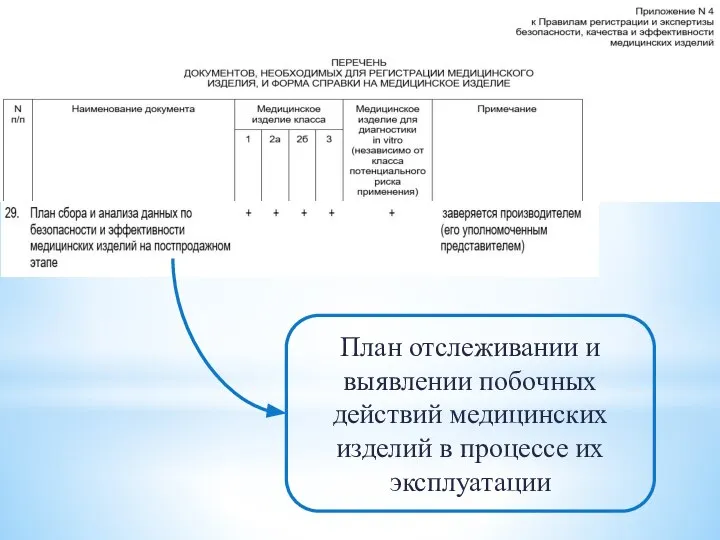
План отслеживании и выявлении побочных действий медицинских изделий в процессе их
План отслеживании и выявлении побочных действий медицинских изделий в процессе их

«Особые»
медицинские изделия
Комплекты, наборы МИ, в состав которых входят уже зарегистрированные МИ
(аптечка,
«Особые»
медицинские изделия
Комплекты, наборы МИ, в состав которых входят уже зарегистрированные МИ
(аптечка,

Источники информации о неблагоприятных событиях
?
FDA, США
MHRA, Великобритания
Health Canada, Канада
Swiss medic, Швейцария
Источники информации о неблагоприятных событиях
?
FDA, США
MHRA, Великобритания
Health Canada, Канада
Swiss medic, Швейцария

Источники информации о неблагоприятных событиях
Источники информации о неблагоприятных событиях
 Вместе против коррупции
Вместе против коррупции Крупнейший оператор кредитных юристов в России Эскалат
Крупнейший оператор кредитных юристов в России Эскалат Брак и семья
Брак и семья Организация хранения документов Архивного фонда Российской Федерации и других архивных документов
Организация хранения документов Архивного фонда Российской Федерации и других архивных документов Национальная политика в области лекарственных средств
Национальная политика в области лекарственных средств Глава государства
Глава государства Экстренные службы города
Экстренные службы города Торжество или равенство Устава Северодвинска
Торжество или равенство Устава Северодвинска Дисциплинарная ответственность прокурорских работников. (Тема 4)
Дисциплинарная ответственность прокурорских работников. (Тема 4) Правовое государство
Правовое государство Объекты гражданских правоотношений
Объекты гражданских правоотношений Межведомственное взаимодействие органов и учреждений системы профилактики безнадзорности и правонарушений несовершеннолетних
Межведомственное взаимодействие органов и учреждений системы профилактики безнадзорности и правонарушений несовершеннолетних Система государственного управления
Система государственного управления 20130401_rossiya_-_rodina_moya_simvolika
20130401_rossiya_-_rodina_moya_simvolika Сущность и задачи стандартизации. Документы в области стандартизации. Организация работ по стандартизации
Сущность и задачи стандартизации. Документы в области стандартизации. Организация работ по стандартизации Комитет по государственному заказу Санкт-Петербурга
Комитет по государственному заказу Санкт-Петербурга Уголовная ответственность за контрабанду наркотических и психотропных веществ, их аналогов, инструментов и оборудования
Уголовная ответственность за контрабанду наркотических и психотропных веществ, их аналогов, инструментов и оборудования Организация и законодательная основа таможенного дела в российской федерации
Организация и законодательная основа таможенного дела в российской федерации Служба в органах и организациях прокуратуры
Служба в органах и организациях прокуратуры 2022_TP_Tema_1_1_TP_v_EAES
2022_TP_Tema_1_1_TP_v_EAES Военные угрозы национальной безопасности России и национальная оборона
Военные угрозы национальной безопасности России и национальная оборона Должностное лицо, уполномоченное осуществлять предварительное следствие
Должностное лицо, уполномоченное осуществлять предварительное следствие Нормативно - правовое регулирование в области защиты территорий от ЧС (тема №1)
Нормативно - правовое регулирование в области защиты территорий от ЧС (тема №1) Таможенное обеспечение международных перевозок и экспедирования грузов
Таможенное обеспечение международных перевозок и экспедирования грузов Право собственности. Документы для сделки
Право собственности. Документы для сделки Права на недвижимое имущество. (тема 18)
Права на недвижимое имущество. (тема 18) Правовое регулирование. Статья 70
Правовое регулирование. Статья 70 Брачно-семейные отношения в Древнем Риме
Брачно-семейные отношения в Древнем Риме