- Основы математического моделирования. Расчеты «из первых принципов»

Содержание
- 2. Цель лекции Рассмотреть понятие вычислительная нанотехнология. Определить классификацию методов моделирования в области нанотехнологий. Изучить квантовомеханические расчеты
- 3. Содержание лекции Вычислительная нанотехнология. Классификация методов моделирования в области нанотехнологий. Квантовомеханические расчеты «из первых принципов». Моделирование
- 4. ВЫЧИСЛИТЕЛЬНАЯ НАНОТЕХНОЛОГИЯ
- 5. Предпосылки моделирования в наномире При разработке наноматериалов с заданными свойствами используются в основном экспериментальные методы, что
- 6. Вычислительная нанотехнология Вычислительная нанотехнология ‒ самостоятельный и эффективный метод познания закономерностей наномира, включающий в себя фундаментальные
- 7. Особенности вычислительной нанотехнологии В настоящее время можно теоретически изучать неизвестные кристаллические структуры, кластеры и молекулы, исследовать
- 8. КЛАССИФИКАЦИЯ МЕТОДОВ МОДЕЛИРОВАНИЯ В ОБЛАСТИ НАНОТЕХНОЛОГИЙ
- 9. Группы математических моделей динамики наносистем Методы математического описания динамики взаимодействующих частиц. Математические модели кинетики кластеризации и
- 10. Методы математического описания динамики взаимодействующих частиц Квантовомеханические расчеты «из первых принципов». Моделирование строения многоэлектронных атомов. Моделирование
- 11. Модели кластерных систем Модель роста кластеров в свободном объеме. Модели нуклеации (фазовый переход) и роста кластеров
- 12. Модели транспортно-диффузионного переноса Механизмы переноса и трансформации вещества и энергии в системе. Особенности процессов в неэкстенсивных
- 13. КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ «ИЗ ПЕРВЫХ ПРИНЦИПОВ»
- 14. Расчеты «из первых принципов» Поскольку решить квантовое уравнение Шредингера для системы многих частиц не представляется возможным,
- 15. КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ «ИЗ ПЕРВЫХ ПРИНЦИПОВ» МОДЕЛИРОВАНИЕ СТРОЕНИЯ МНОГОЭЛЕКТРОННЫХ АТОМОВ
- 16. Моделирование строения многоэлектронных атомов Метод Хартри-Фока (одноэлектронное приближение). Теория функционала плотности .
- 17. Метод Хартри-Фока (одноэлектронное приближение) Суть метода: взаимодействие каждого электрона в атоме со всеми остальными заменяется его
- 18. Метод Хартри-Фока (одноэлектронное приближение) Полная энергия электрона, находящегося в поле двух ядер:
- 19. Метод Хартри-Фока (одноэлектронное приближение) При замене в уравнении (1) последнего члена, зависящего от координат двух электронов,
- 20. Метод Хартри-Фока (одноэлектронное приближение) В.А. Фок усовершенствовал метод Хартри, добавив в уравнение (2) дополнительный член, учитывающий
- 21. Особенности: Решение уравнений Хартри-Фока – итерационный способ. Полученные функции представляют в виде таблиц. Существует модификация метода
- 22. Моделирование различных модификации известных углеродных наноструктур. Моделирование влияния межэлектронного взаимодействия на стационарные характеристики резонансно-туннельного диода. Применение:
- 23. Теория функционала плотности Цель метода: Существенное упрощение задачи: при описании электронной подсистемы заменить многоэлектронную волновую функцию
- 24. Теория функционала плотности Энергия атома рассчитывается как сумма его кинетической энергии, представленной в виде функционала электронной
- 25. Применение метода ТФП Моделирование адсорбции различных молекул. Расчетов диэлектрической проницаемости металлических фотонных кристаллов при высоких температурах.
- 26. КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ «ИЗ ПЕРВЫХ ПРИНЦИПОВ» МОДЕЛИРОВАНИЕ МОЛЕКУЛЯРНЫХ СИСТЕМ
- 27. Моделирование молекулярных систем Планетарная модель . Модель ковалентной связи. Приближение Борна-Оппенгеймера. Метод валентных схем. Метод молекулярных
- 28. Особенности химической связи Все теоретические положения о строении молекул и структуре химических связей держатся на трех
- 29. Планетарная модель Одной из первых моделей двухатомной молекулы была своеобразная планетарная модель, в которой электроны, располагаясь
- 30. Планетарная модель Методы квантовой механики, показывают, что действительно в области между ядрами должна быть повышенная плотность
- 31. Модель ковалентной связи Понятие химической связи было сформулировано в работах А.М. Бутлерова. Химическая связь есть взаимодействие
- 32. Модель ковалентной связи Атомы могут объединяться в молекулы, достигая октета валентных электронов путем их совместного использования.
- 33. Примеры простейших моделей Молекулярные модели молекул: а – воды, б – этилена, в – дихлоэтана, г
- 34. Приближение Борна-Оппенгеймера Оператор Гамильтона молекулы с N ядрами и n электронами содержит члены кинетической энергии электронов
- 35. Приближение Борна-Оппенгеймера Масса ядра значительно превышает массу электрона. Скорость движения ядер значительно меньше по сравнению со
- 36. Приближение Борна-Оппенгеймера В рамках квантовой механики такое приближение эквивалентно допущению, что полная волновая функция молекулы может
- 37. Метод валентных схем Эффективный подход в поиске формы волновой функции был предложен в 1927 году В.
- 38. Метод молекулярных орбиталей В 1927-1929 годах Ф. Хунд, Дж. Леннард-Джонс и Р.С. Малликен развили идею нового
- 39. Метод молекулярных орбиталей В наиболее простой форме метод МО включает следующие основные положения и допущения: рассматриваются
- 40. Сравнение метода ВС и МО Применимость метода ВС только к ограниченному классу молекул. В методе ВС
- 41. Модель поверхности потенциальной энергии В основе фундаментального понятия о поверхности потенциальной энергии лежит приближение Борна-Оппенгеймера, позволяющее
- 42. Модель поверхности потенциальной энергии Примеры форм простейших поверхностей потенциальной энергии, описываемых только двумя координатами q1 и
- 43. Модель поверхности потенциальной энергии В терминах поверхности потенциальной энергии химическая реакция означает переход молекулярной системы из
- 44. КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ «ИЗ ПЕРВЫХ ПРИНЦИПОВ» МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
- 45. Межмолекулярные взаимодействия Межмолекулярные взаимодействия (от десятых до десятка кДж/моль). Химическая связь в молекуле (десятков и сотен
- 46. Свойства межмолекулярных сил На очень большом расстоянии молекулы не взаимодействуют, т.е. гораздо быстрее убывают с расстоянием,
- 47. Диполь-дипольное приближение Идея: каждая молекула создает вокруг себя внешнее электростатическое поле. Это поле всегда может быть
- 48. Диполь-дипольное приближение В классическом понимании дипольный момент это сумма произведений всех радиус-векторов ядер на их заряд.
- 49. Диполь-дипольное приближение Особенности: Приближение является хорошим лишь при больших расстояниях между взаимодействующими молекулами. Все три вида
- 50. Потенциалы взаимодействия частиц Потенциалы межатомного и межмолекулярного взаимодействия предназначены для получения статистически усредненного представления таких сил
- 51. Потенциалы взаимодействия частиц Существует полная база данных по потенциалам межатомного и межмолекулярного взаимодействий для весьма простых
- 52. Потенциал Леннарда-Джонса Данный потенциал является одним из наиболее часто используемых потенциалов. Первоначально этот потенциал был предложен
- 53. Парный потенциал Приближение парного потенциала позволяет перейти от многомерных измерений поверхности потенциальной энергии к многократному суммированию
- 54. Потенциал Букингема Первоначальный вариант потенциала: ε – глубина минимума энергии; rт – соответствующее значение расстояния r
- 55. Модифицированный потенциал Букингема Устраняя член с обратной 8-й степенью, приходим к более простому виду потенциала Букингема:
- 56. Потенциал Морзе Для вычисления энергетических уровней двухатомной молекулы Морзе предложил потенциал взаимодействия, который должен удовлетворять следующим
- 57. Потенциал Морзе Применения: изучение динамики решетки; исследование структуры дефектов в металлах; при расчете упругих свойств металлов;
- 58. Псевдопотенциал Методология псевдопотенциалов опирается на приближение малого остова, согласно которому ионные остовы (ядра с электронами внутренних
- 59. Псевдопотенциал Особенности: Ограниченность метода в силу сделанных допущений: отсутствия перекрытия ионных остовов и постоянства объема системы.
- 60. Многочастичные потенциалы Используются для описания взаимодействия в материалах с ковалентными связями или явлений с высоким энергетическим
- 61. Способы перехода к многочастичным потенциалам Добавление слагаемого в виде функционала электронной плотности данного атома в локальном
- 62. Потенциалы модели «погруженного» атома Многочастичные потенциалы МПА обеспечивают моделирование образования связей в металлических кластерах. Они созданы
- 63. Потенциалы Финниса-Синклера Многочастичные потенциалы Финниса и Синклера были разработаны для моделирования энергетического состояния переходных металлов. где
- 64. Потенциалы Саттона-Чена Дальнодействующие многочастичные потенциалы Саттона и Чена описывают энергетические свойства десяти ГЦК элементарных металлов. По
- 65. Потенциалы Рафии-Табара и Саттона Указанные потенциалы применяются для моделирования энергетического состояния металлических сплавов, и в частности
- 66. Потенциалы Меррелла-Моттрама Многочастичные потенциалы Меррелла-Моттрама являются примером потенциалов кластерных систем и состоят из сумм эффективных потенциалов
- 67. Потенциалы Терсоффа Многочастичные потенциалы Терсоффа предназначены для описания ковалентных взаимодействий атомных пар С-С, Si-Si и C-Si.
- 68. Заключение и выводы Рассмотрено понятие вычислительная нанотехнология. Определена классификация методов моделирования в области нанотехнологий. Изучены квантовомеханические
- 70. Скачать презентацию

Цель лекции
Рассмотреть понятие вычислительная нанотехнология.
Определить классификацию методов моделирования в области нанотехнологий.
Изучить
Цель лекции
Рассмотреть понятие вычислительная нанотехнология.
Определить классификацию методов моделирования в области нанотехнологий.
Изучить

Содержание лекции
Вычислительная нанотехнология.
Классификация методов моделирования в области нанотехнологий.
Квантовомеханические расчеты «из первых
Содержание лекции
Вычислительная нанотехнология.
Классификация методов моделирования в области нанотехнологий.
Квантовомеханические расчеты «из первых

ВЫЧИСЛИТЕЛЬНАЯ НАНОТЕХНОЛОГИЯ
ВЫЧИСЛИТЕЛЬНАЯ НАНОТЕХНОЛОГИЯ

Предпосылки моделирования в наномире
При разработке наноматериалов с заданными свойствами используются в
Предпосылки моделирования в наномире
При разработке наноматериалов с заданными свойствами используются в

Вычислительная нанотехнология
Вычислительная нанотехнология ‒ самостоятельный и эффективный метод познания закономерностей наномира,
Вычислительная нанотехнология
Вычислительная нанотехнология ‒ самостоятельный и эффективный метод познания закономерностей наномира,

Особенности вычислительной нанотехнологии
В настоящее время можно теоретически изучать неизвестные кристаллические структуры,
Особенности вычислительной нанотехнологии
В настоящее время можно теоретически изучать неизвестные кристаллические структуры,

КЛАССИФИКАЦИЯ МЕТОДОВ МОДЕЛИРОВАНИЯ В ОБЛАСТИ НАНОТЕХНОЛОГИЙ
КЛАССИФИКАЦИЯ МЕТОДОВ МОДЕЛИРОВАНИЯ В ОБЛАСТИ НАНОТЕХНОЛОГИЙ

Группы математических моделей динамики наносистем
Методы математического описания динамики взаимодействующих частиц.
Математические модели
Группы математических моделей динамики наносистем
Методы математического описания динамики взаимодействующих частиц.
Математические модели

Методы математического описания динамики взаимодействующих частиц
Квантовомеханические расчеты «из первых принципов».
Моделирование строения
Методы математического описания динамики взаимодействующих частиц
Квантовомеханические расчеты «из первых принципов».
Моделирование строения

Модели кластерных систем
Модель роста кластеров в свободном объеме.
Модели нуклеации (фазовый переход)
Модели кластерных систем
Модель роста кластеров в свободном объеме.
Модели нуклеации (фазовый переход)

Модели транспортно-диффузионного переноса
Механизмы переноса и трансформации вещества и энергии в системе.
Особенности
Модели транспортно-диффузионного переноса
Механизмы переноса и трансформации вещества и энергии в системе.
Особенности

КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»

Расчеты «из первых принципов»
Поскольку решить квантовое уравнение Шредингера для системы
Расчеты «из первых принципов»
Поскольку решить квантовое уравнение Шредингера для системы

КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
МОДЕЛИРОВАНИЕ СТРОЕНИЯ МНОГОЭЛЕКТРОННЫХ АТОМОВ
КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
МОДЕЛИРОВАНИЕ СТРОЕНИЯ МНОГОЭЛЕКТРОННЫХ АТОМОВ

Моделирование строения многоэлектронных атомов
Метод Хартри-Фока (одноэлектронное приближение).
Теория функционала плотности .
Моделирование строения многоэлектронных атомов
Метод Хартри-Фока (одноэлектронное приближение).
Теория функционала плотности .

Метод Хартри-Фока
(одноэлектронное приближение)
Суть метода: взаимодействие каждого электрона в атоме со
Метод Хартри-Фока
(одноэлектронное приближение)
Суть метода: взаимодействие каждого электрона в атоме со
Метод Хартри-Фока
(одноэлектронное приближение)
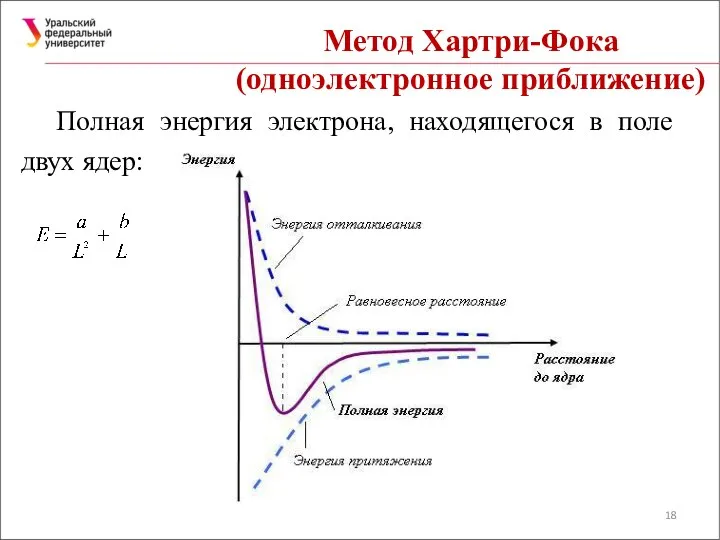
Полная энергия электрона, находящегося в поле двух ядер:
Метод Хартри-Фока
(одноэлектронное приближение)
Полная энергия электрона, находящегося в поле двух ядер:

Метод Хартри-Фока
(одноэлектронное приближение)
При замене в уравнении (1) последнего члена, зависящего
Метод Хартри-Фока
(одноэлектронное приближение)
При замене в уравнении (1) последнего члена, зависящего

Метод Хартри-Фока
(одноэлектронное приближение)
В.А. Фок усовершенствовал метод Хартри, добавив в уравнение
Метод Хартри-Фока
(одноэлектронное приближение)
В.А. Фок усовершенствовал метод Хартри, добавив в уравнение

Особенности:
Решение уравнений Хартри-Фока – итерационный способ.
Полученные функции представляют в виде таблиц.
Существует
Особенности:
Решение уравнений Хартри-Фока – итерационный способ.
Полученные функции представляют в виде таблиц.
Существует
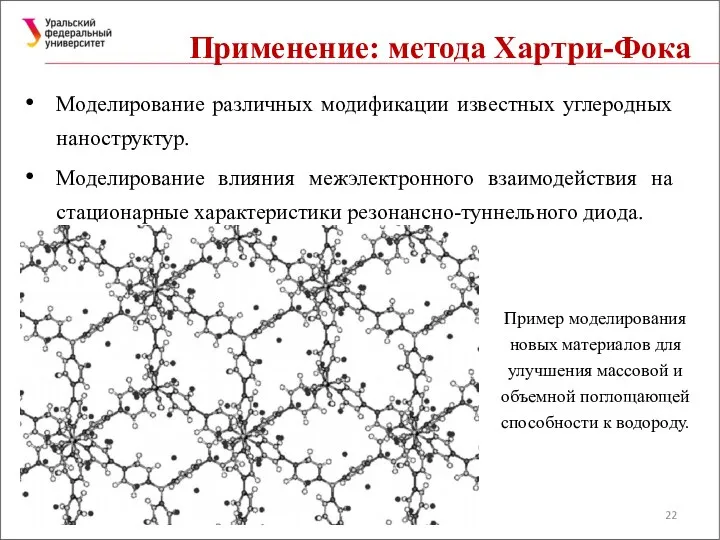
Моделирование различных модификации известных углеродных наноструктур.
Моделирование влияния межэлектронного взаимодействия на стационарные
Моделирование различных модификации известных углеродных наноструктур.
Моделирование влияния межэлектронного взаимодействия на стационарные

Теория функционала плотности
Цель метода: Существенное упрощение задачи: при описании электронной
Теория функционала плотности
Цель метода: Существенное упрощение задачи: при описании электронной

Теория функционала плотности
Энергия атома рассчитывается как сумма его кинетической энергии,
Теория функционала плотности
Энергия атома рассчитывается как сумма его кинетической энергии,
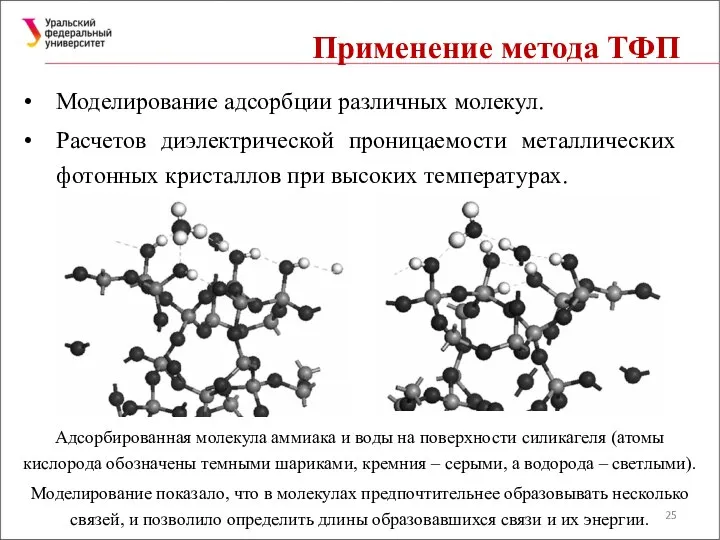
Применение метода ТФП
Моделирование адсорбции различных молекул.
Расчетов диэлектрической проницаемости металлических фотонных
Применение метода ТФП
Моделирование адсорбции различных молекул.
Расчетов диэлектрической проницаемости металлических фотонных

КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
МОДЕЛИРОВАНИЕ МОЛЕКУЛЯРНЫХ СИСТЕМ
КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
МОДЕЛИРОВАНИЕ МОЛЕКУЛЯРНЫХ СИСТЕМ

Моделирование молекулярных систем
Планетарная модель .
Модель ковалентной связи.
Приближение Борна-Оппенгеймера.
Метод валентных схем.
Метод
Моделирование молекулярных систем
Планетарная модель .
Модель ковалентной связи.
Приближение Борна-Оппенгеймера.
Метод валентных схем.
Метод

Особенности химической связи
Все теоретические положения о строении молекул и структуре химических
Особенности химической связи
Все теоретические положения о строении молекул и структуре химических

Планетарная модель
Одной из первых моделей двухатомной молекулы была своеобразная планетарная
Планетарная модель
Одной из первых моделей двухатомной молекулы была своеобразная планетарная

Планетарная модель
Методы квантовой механики, показывают, что действительно в области между
Планетарная модель
Методы квантовой механики, показывают, что действительно в области между

Модель ковалентной связи
Понятие химической связи было сформулировано в работах А.М. Бутлерова.
Модель ковалентной связи
Понятие химической связи было сформулировано в работах А.М. Бутлерова.

Модель ковалентной связи
Атомы могут объединяться в молекулы, достигая октета валентных электронов
Модель ковалентной связи
Атомы могут объединяться в молекулы, достигая октета валентных электронов
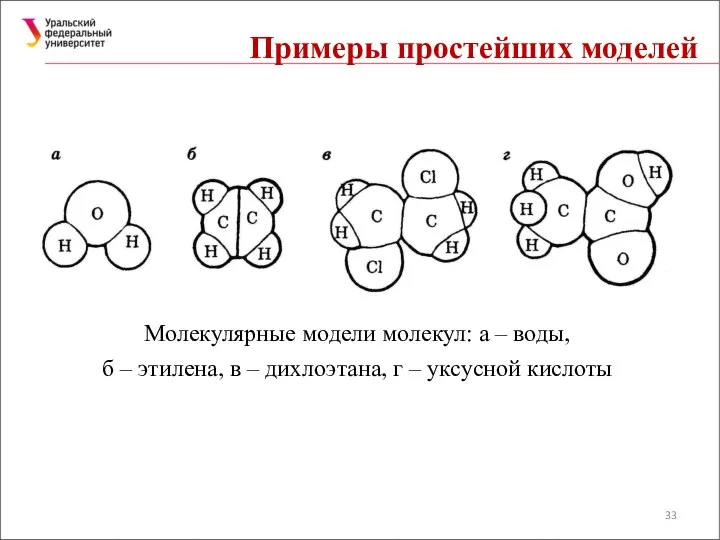
Примеры простейших моделей
Молекулярные модели молекул: а – воды, б – этилена,
Примеры простейших моделей
Молекулярные модели молекул: а – воды, б – этилена,

Приближение Борна-Оппенгеймера
Оператор Гамильтона молекулы с N ядрами и n электронами содержит
Приближение Борна-Оппенгеймера
Оператор Гамильтона молекулы с N ядрами и n электронами содержит

Приближение Борна-Оппенгеймера
Масса ядра значительно превышает массу электрона.
Скорость движения ядер значительно меньше
Приближение Борна-Оппенгеймера
Масса ядра значительно превышает массу электрона.
Скорость движения ядер значительно меньше

Приближение Борна-Оппенгеймера
В рамках квантовой механики такое приближение эквивалентно допущению, что полная
Приближение Борна-Оппенгеймера
В рамках квантовой механики такое приближение эквивалентно допущению, что полная

Метод валентных схем
Эффективный подход в поиске формы волновой функции был предложен
Метод валентных схем
Эффективный подход в поиске формы волновой функции был предложен

Метод молекулярных орбиталей
В 1927-1929 годах Ф. Хунд, Дж. Леннард-Джонс и Р.С.
Метод молекулярных орбиталей
В 1927-1929 годах Ф. Хунд, Дж. Леннард-Джонс и Р.С.

Метод молекулярных орбиталей
В наиболее простой форме метод МО включает следующие основные
Метод молекулярных орбиталей
В наиболее простой форме метод МО включает следующие основные

Сравнение метода ВС и МО
Применимость метода ВС только к ограниченному классу
Сравнение метода ВС и МО
Применимость метода ВС только к ограниченному классу

Модель поверхности потенциальной энергии
В основе фундаментального понятия о поверхности потенциальной энергии
Модель поверхности потенциальной энергии
В основе фундаментального понятия о поверхности потенциальной энергии
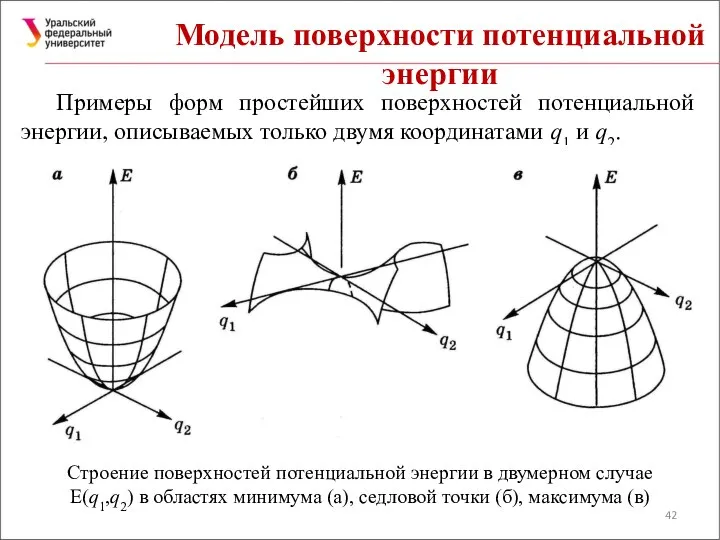
Модель поверхности потенциальной энергии
Примеры форм простейших поверхностей потенциальной энергии, описываемых только
Модель поверхности потенциальной энергии
Примеры форм простейших поверхностей потенциальной энергии, описываемых только
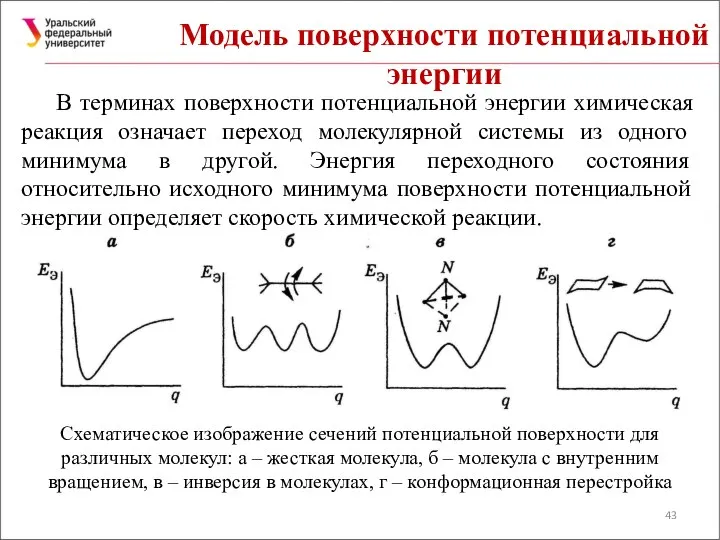
Модель поверхности потенциальной энергии
В терминах поверхности потенциальной энергии химическая реакция означает
Модель поверхности потенциальной энергии
В терминах поверхности потенциальной энергии химическая реакция означает

КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
КВАНТОВОМЕХАНИЧЕСКИЕ РАСЧЕТЫ
«ИЗ ПЕРВЫХ ПРИНЦИПОВ»
МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ

Межмолекулярные взаимодействия
Межмолекулярные взаимодействия (от десятых до десятка кДж/моль). Химическая связь в
Межмолекулярные взаимодействия
Межмолекулярные взаимодействия (от десятых до десятка кДж/моль). Химическая связь в

Свойства межмолекулярных сил
На очень большом расстоянии молекулы не взаимодействуют, т.е. гораздо
Свойства межмолекулярных сил
На очень большом расстоянии молекулы не взаимодействуют, т.е. гораздо

Диполь-дипольное приближение
Идея: каждая молекула создает вокруг себя внешнее электростатическое поле. Это
Диполь-дипольное приближение
Идея: каждая молекула создает вокруг себя внешнее электростатическое поле. Это

Диполь-дипольное приближение
В классическом понимании дипольный момент это сумма произведений всех радиус-векторов
Диполь-дипольное приближение
В классическом понимании дипольный момент это сумма произведений всех радиус-векторов

Диполь-дипольное приближение
Особенности:
Приближение является хорошим лишь при больших расстояниях между взаимодействующими молекулами.
Все
Диполь-дипольное приближение
Особенности:
Приближение является хорошим лишь при больших расстояниях между взаимодействующими молекулами.
Все

Потенциалы взаимодействия частиц
Потенциалы межатомного и межмолекулярного взаимодействия предназначены для получения статистически
Потенциалы взаимодействия частиц
Потенциалы межатомного и межмолекулярного взаимодействия предназначены для получения статистически

Потенциалы взаимодействия частиц
Существует полная база данных по потенциалам межатомного и межмолекулярного
Потенциалы взаимодействия частиц
Существует полная база данных по потенциалам межатомного и межмолекулярного

Потенциал Леннарда-Джонса
Данный потенциал является одним из наиболее часто используемых потенциалов.
Первоначально этот
Потенциал Леннарда-Джонса
Данный потенциал является одним из наиболее часто используемых потенциалов.
Первоначально этот

Парный потенциал
Приближение парного потенциала позволяет перейти от многомерных измерений поверхности потенциальной
Парный потенциал
Приближение парного потенциала позволяет перейти от многомерных измерений поверхности потенциальной

Потенциал Букингема
Первоначальный вариант потенциала:
ε – глубина минимума энергии; rт – соответствующее
Потенциал Букингема
Первоначальный вариант потенциала:
ε – глубина минимума энергии; rт – соответствующее

Модифицированный потенциал Букингема
Устраняя член с обратной 8-й степенью, приходим к более
Модифицированный потенциал Букингема
Устраняя член с обратной 8-й степенью, приходим к более

Потенциал Морзе
Для вычисления энергетических уровней двухатомной молекулы Морзе предложил потенциал взаимодействия,
Потенциал Морзе
Для вычисления энергетических уровней двухатомной молекулы Морзе предложил потенциал взаимодействия,

Потенциал Морзе
Применения:
изучение динамики решетки;
исследование структуры дефектов в металлах;
при расчете упругих свойств
Потенциал Морзе
Применения:
изучение динамики решетки;
исследование структуры дефектов в металлах;
при расчете упругих свойств

Псевдопотенциал
Методология псевдопотенциалов опирается на приближение малого остова, согласно которому ионные остовы
Псевдопотенциал
Методология псевдопотенциалов опирается на приближение малого остова, согласно которому ионные остовы

Псевдопотенциал
Особенности:
Ограниченность метода в силу сделанных допущений: отсутствия перекрытия ионных остовов и
Псевдопотенциал
Особенности:
Ограниченность метода в силу сделанных допущений: отсутствия перекрытия ионных остовов и

Многочастичные потенциалы
Используются для описания взаимодействия в материалах с ковалентными связями или
Многочастичные потенциалы
Используются для описания взаимодействия в материалах с ковалентными связями или

Способы перехода к многочастичным потенциалам
Добавление слагаемого в виде функционала электронной плотности
Способы перехода к многочастичным потенциалам
Добавление слагаемого в виде функционала электронной плотности

Потенциалы модели «погруженного» атома
Многочастичные потенциалы МПА обеспечивают моделирование образования связей в
Потенциалы модели «погруженного» атома
Многочастичные потенциалы МПА обеспечивают моделирование образования связей в

Потенциалы Финниса-Синклера
Многочастичные потенциалы Финниса и Синклера были разработаны для моделирования энергетического
Потенциалы Финниса-Синклера
Многочастичные потенциалы Финниса и Синклера были разработаны для моделирования энергетического

Потенциалы Саттона-Чена
Дальнодействующие многочастичные потенциалы Саттона и Чена описывают энергетические свойства десяти
Потенциалы Саттона-Чена
Дальнодействующие многочастичные потенциалы Саттона и Чена описывают энергетические свойства десяти

Потенциалы Рафии-Табара и Саттона
Указанные потенциалы применяются для моделирования энергетического состояния металлических
Потенциалы Рафии-Табара и Саттона
Указанные потенциалы применяются для моделирования энергетического состояния металлических

Потенциалы Меррелла-Моттрама
Многочастичные потенциалы Меррелла-Моттрама являются примером потенциалов кластерных систем и состоят
Потенциалы Меррелла-Моттрама
Многочастичные потенциалы Меррелла-Моттрама являются примером потенциалов кластерных систем и состоят

Потенциалы Терсоффа
Многочастичные потенциалы Терсоффа предназначены для описания ковалентных взаимодействий атомных пар
Потенциалы Терсоффа
Многочастичные потенциалы Терсоффа предназначены для описания ковалентных взаимодействий атомных пар

Заключение и выводы
Рассмотрено понятие вычислительная нанотехнология.
Определена классификация методов моделирования в области
Заключение и выводы
Рассмотрено понятие вычислительная нанотехнология.
Определена классификация методов моделирования в области
 Решение дробных рациональных уравнений. 8 класс
Решение дробных рациональных уравнений. 8 класс Образование чисел второго десятка. 1 класс
Образование чисел второго десятка. 1 класс Измерительный инструмент
Измерительный инструмент Современные методы статистического анализа
Современные методы статистического анализа Углы, вписанные в окружность
Углы, вписанные в окружность Задачи по геометрия на объем
Задачи по геометрия на объем Знаменитости в математике
Знаменитости в математике Счёт в пределах 1000. Часть 2
Счёт в пределах 1000. Часть 2 Сумма углов треугольника
Сумма углов треугольника Множества. Логические символы. (Лекция 1)
Множества. Логические символы. (Лекция 1) Лесная школа. Игра-тренажёр. Математика для детей 5-7 лет
Лесная школа. Игра-тренажёр. Математика для детей 5-7 лет Платоновы тела. Правильные выпуклые многогранники
Платоновы тела. Правильные выпуклые многогранники Сложение натуральных чисел и его свойства
Сложение натуральных чисел и его свойства Комбинаторика. Комбинаторные конструкции
Комбинаторика. Комбинаторные конструкции Машинная арифметика в рациональных чисел. Лекция №6
Машинная арифметика в рациональных чисел. Лекция №6 Выпуклые четырёхугольники. Специфика параллелограммов. Специфика трапеций
Выпуклые четырёхугольники. Специфика параллелограммов. Специфика трапеций Веселый счет для детей 6-7 лет
Веселый счет для детей 6-7 лет Задачи №№ 1- 8
Задачи №№ 1- 8 Площади фигур
Площади фигур Презентация на тему "Упрощение выражений"
Презентация на тему "Упрощение выражений" Решение задач на смеси, сплавы, растворы.
Решение задач на смеси, сплавы, растворы.  Решение алгебраических уравнений Выполнил: Нелюбин Алексей 9 «В» класс Школа№3 г. Свирск
Решение алгебраических уравнений Выполнил: Нелюбин Алексей 9 «В» класс Школа№3 г. Свирск Числовые, функциональные и степенные ряды
Числовые, функциональные и степенные ряды Умножение и деление. Все действия натуральных чисел с натуральными числами. Метеоры и метеориты
Умножение и деление. Все действия натуральных чисел с натуральными числами. Метеоры и метеориты Деление с остатком
Деление с остатком Рисуем птичек из геометрических фигур
Рисуем птичек из геометрических фигур Презентация по математике "Слово-термин «Логарифм»" - скачать
Презентация по математике "Слово-термин «Логарифм»" - скачать  Конечно-разностные методы решения систем уравнений, описывающих нестационарные режимы работы теплообменника
Конечно-разностные методы решения систем уравнений, описывающих нестационарные режимы работы теплообменника