- Липидозы

Содержание
- 2. СОДЕРЖАНИЕ Определение заболевания – 3 слайд Классификация заболевания – 4 слайд Болезнь Тея-Сакса – 5 слайд
- 3. ОПРЕДЕЛЕНИЕ ЗАБОЛЕВАНИЯ Липидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов
- 4. КЛАССИФИКАЦИЯ ЗАБОЛЕВАНИЯ: Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений, состоящих из углеводов
- 5. БОЛЕЗНЬ ТЕЯ-САКСА Болезнь Тея-Сакса – это генетическое заболевание, характеризующееся недостаточностью фермента гексозаминидазы А, скоплением липоидных макромолекул
- 6. Механизм развития: Наследуется по аутосомно-рецессивному типу Мутация гена HEXA, который расположен на 15 хромосоме Нарушение моторных
- 7. БОЛЕЗНЬ НИМАНА-ПИКА Болезнь Нимана-Пика (сфингомиелиноз) – это наследственное заболевание, связанное с избыточным накоплением жиров в различных
- 8. Механизм развития: Наследуется по аутосомно-рецессивному типу Мутация гена SMPD1, NPC1 и NPC2. Нарушение расщепления сфингомиелина на
- 9. ФЕНОТИП БОЛЬНОГО https://fb.ru/article/166418/bolezn-teya-saksa-redkoe-nasledstvennoe-zabolevanie http://www.urologi.ru/books/3_178.html
- 10. БОЛЕЗНЬ ГОШЕ Болезнь Гоше – это генетическое заболевание, характеризующееся нарушением липидного обмена, недостаточностью лизосомальных ферментов, накоплением
- 11. Механизм развития: Наследуется по аутосомно-рецессивному типу Мутация гена GBA Нарушение синтеза фермента глюкоцереброзидазы Нарушение процесса расщепления
- 12. БОЛЕЗНЬ ФАБРИ Болезнь Фабри – наследственное заболевание, при котором дефект в структуре генов обуславливает недостаточную активность
- 13. Механизм развития: Наследуется по рецессивному Х-сцепленному типу Мутация гена GLA на хромосоме Xq 22.1 Нарушение метаболизма
- 14. https://luxmama.ru/bolezn-goshe-foto-lechenie-simptomyi https://healthworker.ru/bolezn-fabri/
- 15. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ: Клиническая картина болезни отражает процессы поражения ЦНС. В начальной стадии: Первые симптомы становятся заметными
- 16. ДИАГНОСТИКА Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи,
- 17. Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают
- 19. Скачать презентацию

СОДЕРЖАНИЕ
Определение заболевания – 3 слайд
Классификация заболевания – 4 слайд
Болезнь Тея-Сакса
СОДЕРЖАНИЕ
Определение заболевания – 3 слайд
Классификация заболевания – 4 слайд
Болезнь Тея-Сакса

ОПРЕДЕЛЕНИЕ ЗАБОЛЕВАНИЯ
Липидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов
ОПРЕДЕЛЕНИЕ ЗАБОЛЕВАНИЯ
Липидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов

КЛАССИФИКАЦИЯ ЗАБОЛЕВАНИЯ:
Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений,
КЛАССИФИКАЦИЯ ЗАБОЛЕВАНИЯ:
Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений,

БОЛЕЗНЬ ТЕЯ-САКСА
Болезнь Тея-Сакса – это генетическое заболевание, характеризующееся недостаточностью фермента гексозаминидазы А,
БОЛЕЗНЬ ТЕЯ-САКСА
Болезнь Тея-Сакса – это генетическое заболевание, характеризующееся недостаточностью фермента гексозаминидазы А,
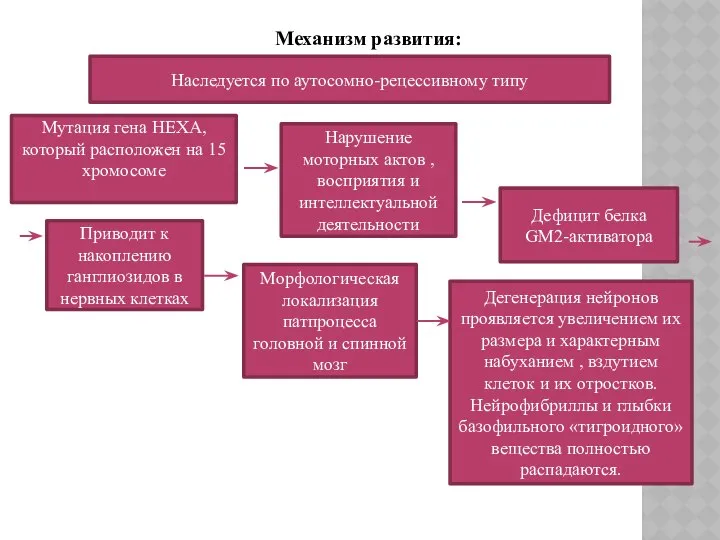
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена HEXA, который расположен на
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена HEXA, который расположен на

БОЛЕЗНЬ НИМАНА-ПИКА
Болезнь Нимана-Пика (сфингомиелиноз) – это наследственное заболевание, связанное с избыточным
БОЛЕЗНЬ НИМАНА-ПИКА
Болезнь Нимана-Пика (сфингомиелиноз) – это наследственное заболевание, связанное с избыточным
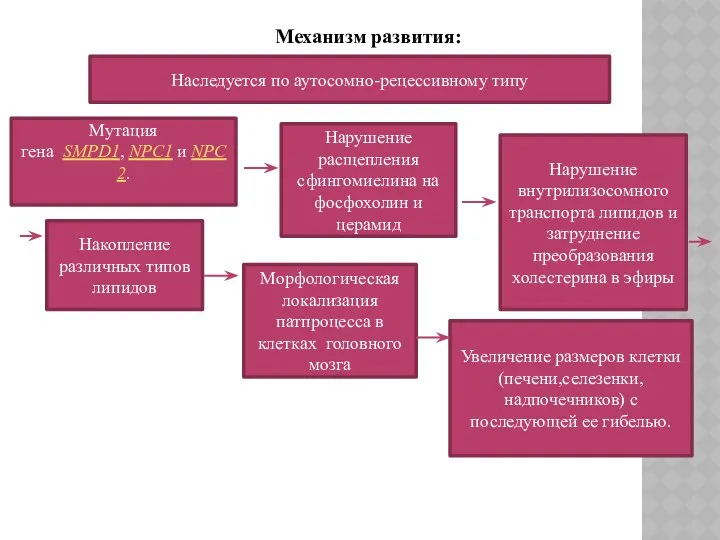
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена SMPD1, NPC1 и NPC2.
Нарушение расщепления сфингомиелина
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена SMPD1, NPC1 и NPC2.
Нарушение расщепления сфингомиелина
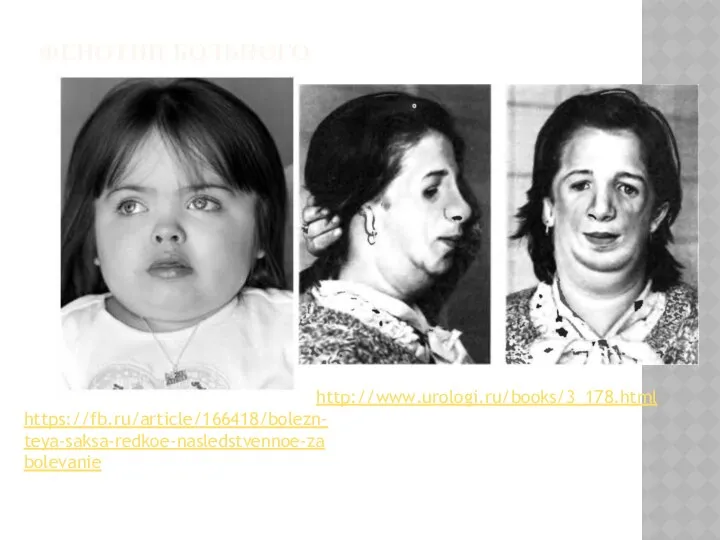
ФЕНОТИП БОЛЬНОГО
https://fb.ru/article/166418/bolezn-teya-saksa-redkoe-nasledstvennoe-zabolevanie
http://www.urologi.ru/books/3_178.html
ФЕНОТИП БОЛЬНОГО
https://fb.ru/article/166418/bolezn-teya-saksa-redkoe-nasledstvennoe-zabolevanie
http://www.urologi.ru/books/3_178.html

БОЛЕЗНЬ ГОШЕ
Болезнь Гоше – это генетическое заболевание, характеризующееся нарушением липидного обмена, недостаточностью
БОЛЕЗНЬ ГОШЕ
Болезнь Гоше – это генетическое заболевание, характеризующееся нарушением липидного обмена, недостаточностью
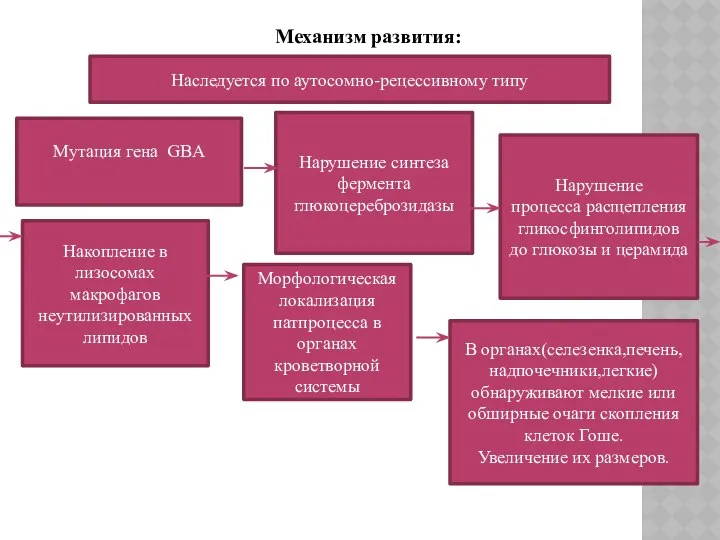
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена GBA
Нарушение синтеза фермента глюкоцереброзидазы
Нарушение
процесса
Механизм развития:
Наследуется по аутосомно-рецессивному типу
Мутация гена GBA
Нарушение синтеза фермента глюкоцереброзидазы
Нарушение
процесса

БОЛЕЗНЬ ФАБРИ
Болезнь Фабри – наследственное заболевание, при котором дефект в структуре генов
БОЛЕЗНЬ ФАБРИ
Болезнь Фабри – наследственное заболевание, при котором дефект в структуре генов
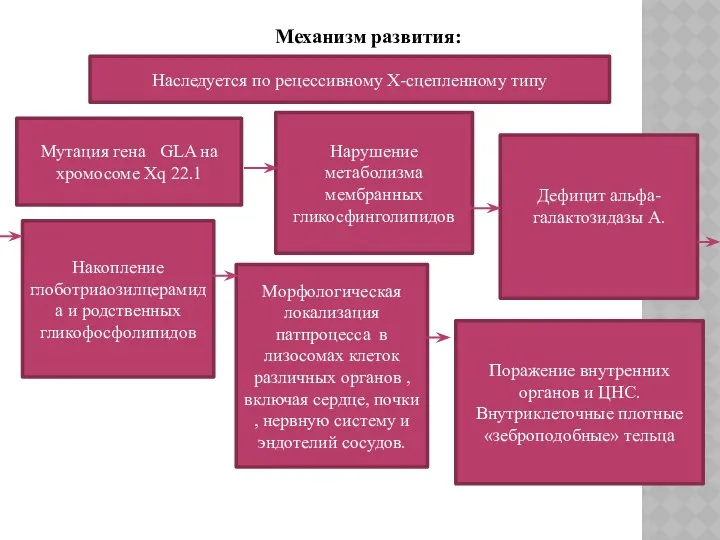
Механизм развития:
Наследуется по рецессивному Х-сцепленному типу
Мутация гена GLA на
хромосоме
Механизм развития:
Наследуется по рецессивному Х-сцепленному типу
Мутация гена GLA на
хромосоме
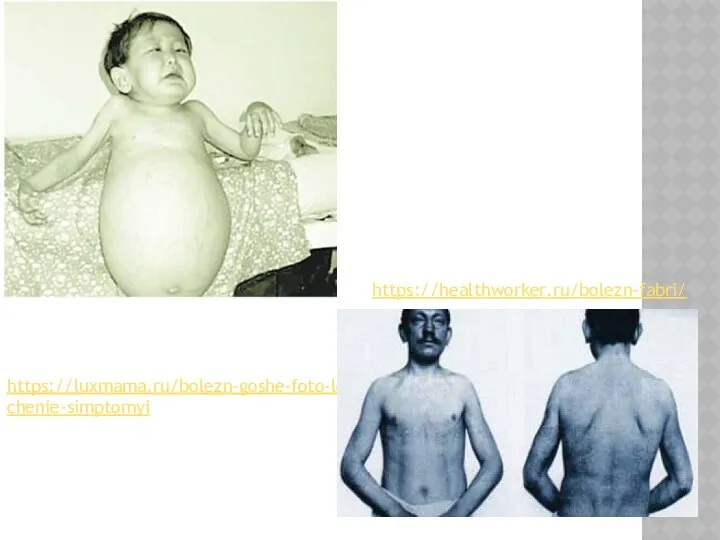
https://luxmama.ru/bolezn-goshe-foto-lechenie-simptomyi
https://healthworker.ru/bolezn-fabri/
https://luxmama.ru/bolezn-goshe-foto-lechenie-simptomyi
https://healthworker.ru/bolezn-fabri/

КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ:
Клиническая картина болезни отражает процессы поражения ЦНС.
В начальной стадии:
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ:
Клиническая картина болезни отражает процессы поражения ЦНС.
В начальной стадии:

ДИАГНОСТИКА
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением
ДИАГНОСТИКА
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением

Терапия данной группы заболеваний – сложная задача для врачей разных специальностей.
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей.
 Влияние COVID-19 на хронические заболевания
Влияние COVID-19 на хронические заболевания Зоонозные заболевания
Зоонозные заболевания Обследование и синдромы при заболеваниях почек
Обследование и синдромы при заболеваниях почек Жүйке жүйесінің ауруларындағы емдік дене шынықтыру және емдік тағам
Жүйке жүйесінің ауруларындағы емдік дене шынықтыру және емдік тағам СРС: Эпидемиологическая характеристика и профилактика нозокомиального туберкулеза
СРС: Эпидемиологическая характеристика и профилактика нозокомиального туберкулеза МСЭ при аневризме сердца
МСЭ при аневризме сердца Малярия
Малярия Кетогенная диета как альтернативный метод лечения фармакорезистентной эпилепсии
Кетогенная диета как альтернативный метод лечения фармакорезистентной эпилепсии Профилактика сахарного диабета
Профилактика сахарного диабета Лекарственные средства с недоказанной эффективностью
Лекарственные средства с недоказанной эффективностью Что такое персональная эффективность
Что такое персональная эффективность Дети с задержкой психического развития
Дети с задержкой психического развития Сестринское обеспечение хирургических перевязок
Сестринское обеспечение хирургических перевязок Оболочечные проявления ЧМТ
Оболочечные проявления ЧМТ Балалардағы жүрек-қантамыр жүйесі мүшелерінің құрылысының ерекшеліктері
Балалардағы жүрек-қантамыр жүйесі мүшелерінің құрылысының ерекшеліктері Сестринский уход при заболеваниях органов дыхания у гериатрических пациентов
Сестринский уход при заболеваниях органов дыхания у гериатрических пациентов Инфекции МВС. Часть №1
Инфекции МВС. Часть №1 Иммунология на службе здоровья
Иммунология на службе здоровья Интенсивная терапия при острой сердечной и острой сердечно – сосудистой недостаточности
Интенсивная терапия при острой сердечной и острой сердечно – сосудистой недостаточности Здоровьесберегающие физкультурно-оздоровительные технологии в физическом воспитании детей и подростков
Здоровьесберегающие физкультурно-оздоровительные технологии в физическом воспитании детей и подростков Пищевые отравления
Пищевые отравления Coronovirus infection
Coronovirus infection Консервативные методы в гинекологии
Консервативные методы в гинекологии Анодонтия. Первичная адентия
Анодонтия. Первичная адентия Морально-нравственные проблемы фармакологического экспериментирования
Морально-нравственные проблемы фармакологического экспериментирования Ревматические болезни
Ревматические болезни А дәрумені (ретинол)
А дәрумені (ретинол) Диагностика сахарного диабета
Диагностика сахарного диабета