- Міопатії. Невральні аміотрофії. Міотонія Томпсона

Содержание
- 2. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Група захворювань (якщо мова йде про первинну міопатію), яка не залежить від
- 3. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) З сучасних авторів багато працювали в цій галузі: Белл, Стівенсон, Шаргородський, Мельников,
- 4. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Класифікація міопатій: (Л.О.Бадалян, 1973) I. Первинні міопатії: Юнацька (ювенільна) форма Ерба; Псевдогіпертрофічна
- 5. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Класифікація міопатій: (Л.О.Бадалян, 1973) II. Вторинні міопатії (аміотрофії): Спінальна аміотрофія Вердніга-Гоффманна; Невральна
- 6. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Класифікація міопатій (Л.О.Бадалян, 1973) IV. Міопатичні синдроми (фенокопії ПМД) при : ендокринних
- 7. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Не всі визнають розподіл м'язових дистрофій на первинні (міогенні) та вторинні (невральні),
- 8. ПЕРВИННІ МІОПАТІЇ: Патологічна анатомія. При аутопсії: м'язи стоншені, жовто-сірого кольору в зв'язку з заміщенням м'язових волокон
- 9. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Етіологія і патогенез. Міопатії відносяться до групи спадкових захворювань. Встановлено, що плечо-лопатково-лицева
- 10. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Етіологія і патогенез. Поряд з чіткими спадковими формами міопатій постерігається велика кількість
- 11. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Патогенез ПМД на даний момент повністю не з'ясований. Неврогенна гіпотеза пояснює зміни
- 12. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Патогенез ПМД. При ПМД Дрейфуса (рідкісна форма з контрактурами, дебільністю, без псевдогіпертрофії)
- 13. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
- 14. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Перебіг та симптоми. Головними клінічними симптомами міопатії є: -повільно наростаюча атрофія скелетної
- 15. Міопатія (м'язові атрофії)
- 16. Міопатія (псевдогіпертрофії)
- 17. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Симптоми. Атрофії розвиваються дуже повільно, вражаючи поступово (і зазвичай симетрично) одні м'язові
- 18. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Симптоми. Характерно для міопатій: гладенький полірований лоб, недостатнє змикання очей, малорухливі та
- 19. Міопатія (симптом вигнутої спини та псевдогіпертрофії)
- 20. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
- 21. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
- 22. Симптом «драбинки» - вставання драбинкою
- 23. Симптом Говерса (вставання з підлоги)
- 24. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ) Симптоми. Інтелект не страждає (якщо не брати до уваги окремі випадки форми
- 25. ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
- 26. ОКРЕМІ ФОРМИ МІОПАТІЙ Ювенільна (юнацька) форма Ерба це найбільш аморфна форма, більшість фенокопій припадає саме на
- 27. Міопатія Ерба
- 28. ОКРЕМІ ФОРМИ МІОПАТІЙ Псевдогіпертрофічна форма Дюшена хвороба успадковується за рецесивним типом, зчепленим зі статтю (у 90%
- 29. Міопатія Дюшена
- 30. ОКРЕМІ ФОРМИ МІОПАТІЙ Псевдогіпертрофічна форма Дюшена
- 31. Міопатія Дюшена
- 32. М'ЯЗОВА ДИСТРОФІЯ БЕККЕРА-КІНЕРА Х-зчеплена доброякісна міодистрофія, що була описана в 1955 році; починається в 10-15 років,
- 33. Міопатія Беккера-Кінера
- 34. ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА Захворювання успадковується за аутосомно-домінантним типом з високою пенетрантністю і варіабельною експресивністю; хворіють особи
- 35. ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
- 36. ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА Рідкісні форми: - дистальна; - окулярна; - окулофарінгеальна. ЕМГ вказує на ураження м'язів
- 37. ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
- 38. Диференціальна діагностика міопатій Іноді подібну клінічну картину має хвороба Вердніга-Гоффманна, але для неї характерна невральна форма
- 39. ЛІКУВАННЯ МІОПАТІЙ Лікування міопатії-повинно бути постійним або з короткими перервами. Лікувати необхідно з фахівцями ЛФК та
- 40. ЛІКУВАННЯ МІОПАТІЙ В той же час деякі автори не підтверджують позитивної дії АТФ, метіоніну, вітаміну Е,
- 41. ЛІКУВАННЯ МІОПАТІЙ Зразок схеми курсів лікування ПМД за Бадаляновим Л.О. I курс- АТФ №30, Галантамін №20,
- 42. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Описали Шарко і Марі в 1806 р. (Франція), Тут -
- 43. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Найчастіше успадковується за домінантним типом зі значною пенетрантністю та неповною
- 44. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Патологічна анатомія. Відзначається переродження осьових циліндрів периферичних нервів і нервових
- 45. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Патологічна анатомія.
- 46. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Клініка. Процес, перш за все, вражає стопи, викликаючи їх атрофію,
- 47. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Клініка.
- 48. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Клініка.
- 49. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Клініка.
- 50. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Клініка. Значно рідше першими в процес залучаються верхні кінцівки. Іноді
- 51. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Перебіг захворювання повільний, але неухильний. Хворі довго зберігають працездатність. Діагноз.
- 52. ВТОРИННІ АМІОТРОФІЇ: Невральна м'язова аміотрофія Шарко-Марі-Тута Лікування: вітаміни групи В; АТФ; антихолінестеразні препарати; анаболічні стероїди; Секуренін;
- 53. СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ форма ВЕРДНІГА-ГОФФМАННА Захворювання описав Вердніг в 1891р. Гоффман провів детальне дослідження
- 54. СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ форма ВЕРДНІГА-ГОФФМАННА При вродженій формі: майже відсутнє ворушіння плода, нерухомість дитини
- 55. СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ форма ВЕРДНІГА-ГОФФМАННА
- 56. СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ форма ВЕРДНІГА-ГОФФМАННА Пізня форма. Зазвичай починається у дітей, які до цього
- 57. СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ форма ВЕРДНІГА-ГОФФМАННА Діагноз. Як правило хворі надходять з діагнозом дистрофія або
- 58. СПІНАЛЬНІ АМІОТРОФІЇ ДОРОСЛИХ (хвороба Арана- Дюшена) Це дуже неясний розділ клінічної неврології, однак, такі хворі є
- 59. СПІНАЛЬНІ АМІОТРОФІЇ ДОРОСЛИХ Клініка. Основний симптом захворювання - м'язова атрофія, що починається, зазвичай, з кистей, далі
- 60. СПІНАЛЬНІ АМІОТРОФІЇ ДОРОСЛИХ Таким чином, виділяють 3 форми спінальної аміотрофії: хронічна дистальна; хронічна проксимальна; бульбарно-спінальна аміотрофія
- 61. СПІНАЛЬНІ АМІОТРОФІЇ ДОРОСЛИХ Діагноз. Звичайно, найбільші труднощі виникають при диференціальній діагностиці з іншими спінальними аміотрофіями та
- 62. СПІНАЛЬНІ АМІОТРОФІЇ ДОРОСЛИХ Лікування: антихолінестеразні препарати, стероїди, вітаміни групи В та Е, пірацетам, аміналон, піридітол, електростимуляція.
- 63. МІОТОНІЯ ХВОРОБА ТОМСЕНА Міотонія - особливий стан м'язів, що пояснюється в тому, що м'яз, який перебуває
- 64. МІОТОНІЯ ХВОРОБА ТОМСЕНА За даними А.П.Зінченко з 110 хворих на міотонію тільки у 47% спостерігалися захворювання
- 65. МІОТОНІЯ ХВОРОБА ТОМСЕНА Клініка. Хворий після стану спокою з великими труднощами починає рухатись, а, зробивши крок,
- 66. МІОТОНІЯ ХВОРОБА ТОМСЕНА
- 67. МІОТОНІЯ ХВОРОБА ТОМСЕНА
- 68. МІОТОНІЯ ХВОРОБА ТОМСЕНА
- 69. МІОТОНІЯ ХВОРОБА ТОМСЕНА Хворі відрізняються атлетичною статурою. М'язи їх на дотик щільні та тверді, але сила
- 70. МІОТОНІЯ ХВОРОБА ТОМСЕНА Лікування. Застосовують теплі ванни та інші теплові процедури, помірну ЛФК. Шінявський та Астрейко
- 71. Спадкові захворювання нервової системи (хвороба Штрюмпелля, атаксії, хорея Гентінґтона, хвороба Вільсона-Коновалова) ЗАПОРІЗЬКИЙ ДЕРЖАВНИЙ МЕДИЧНИЙ УНІВЕРСИТЕТ КАФЕДРА
- 72. СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ Пат. анатомія, патогенез Хронічно прогресуюче спадкове захворювання. Вперше описав у 1876 р.
- 73. СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ Клінічна картина: Клініка. Частіше хворіють на 2-му десятиріччі життя, хоча відомі родини,
- 74. СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ Клінічна картина: Так, за даними Ozvath з 267 сімей у 57% були
- 75. СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ Діагноз У типових випадках за наявністю сімейного анамнезу постановка діагнозу утруднень не
- 76. СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ Лікування: Зниження м'язового тонусу: ліорезал в ін'єкціях (Швейцарія), баклофен 1таблетка x 3
- 77. СПАДКОВІ АТАКСІЇ Спадкові атаксії зустрічаються в різних формах і в кожній формі є свої варіанти. Крім
- 78. СІМЕЙНА АТАКСІЯ ФРІДРАЙХА Пат. анатомія, патогенез: Описана цим автором в 1861 р. Відноситься до спадкове захворювання
- 79. СІМЕЙНА АТАКСІЯ ФРІДРАЙХА Клінічна картина: Характерні екстраневральні порушення: А) зміна скелета: кіфосколіоз, стопи з глибоким склепінням
- 80. СІМЕЙНА АТАКСІЯ ФРІДРАЙХА Діагноз: Діагноз в типових випадках не складний, але типові випадки бувають не так
- 81. СІМЕЙНА АТАКСІЯ ФРІДРАЙХА Лікування: Лікування симптоматичне: - Френелевська гімнастика, - Вітаміни групи В, - Препарати типу
- 82. МОЗОЧКОВА МІОКЛОНІЧНА ДИССИНЕРГІЯ ХАНТА Пат. анатомія, клініка, лікування. Описана Хантом в 1921р. Пат анатомія. Дегенеративні зміни
- 83. ХОРЕЯ ГЕНТІНҐТОНА (ХОРЕІЧНА ДЕМЕНЦІЯ) Пат. анатомія, патогенез: Хронічне прогресуюче захворювання, основними ознаками якого є хореїчний гіперкінез
- 84. ХОРЕЯ ГЕНТІНҐТОНА Клініка: Клінічна картина. Гіперкінез проявляється швидкими неритмічними довільними рухами, що безладно виникають в різних
- 85. ХОРЕЯ ГЕНТІНҐТОНА Клініка: Приблизно у 10-15% хворих на ХГ є акінетико-ригідна форма. Хореїчний гіперкінез у них
- 86. ХОРЕЯ ГЕНТІНҐТОНА Клініка: Клініка. Основний симптом - це локомоторна та статична атаксія. Хворі ходять невпевнено, широко
- 87. ХОРЕЯ ГЕНТІНҐТОНА Діагноз: Діагноз і диференційний діагноз. Поєднання хореїформного гіперкінезу з деменцією, пізній початок і неухильно
- 88. ХОРЕЯ ГЕНТІНҐТОНА Лікування: Необхідно: блокувати дофамінергічні рецептори або зменшити вміст дофаміну (у хворих на ХГ він
- 89. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Пат. анатомія, патогенез: У 1883 р. Штромпелль описав захворювання, яке назвав псевдосклероз,
- 90. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Клініка: Клінічна картина. Неврологічні симптоми розвиваються поступово: спочатку порушується м'язовий тонус, потім
- 91. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Клініка: психічні розлади при цій хворобі вельми різноманітні. Хворі мляві і разом
- 92. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Кільце Кайзера-Флейшера
- 93. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Етіологія, пат. анатомія, патогенез, клініка: Крім мозкових явищ важливе місце в клінічній
- 94. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Єтіологія, пат. анатомія, патогенез, клініка: Вочевидь батько і мати є гетерозиготами з
- 95. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Що ж власне передається у спадок? Який патогенез хвороби? При цій хворобі
- 96. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Діагноз: Діагноз. Прогресуюче захворювання, з переважним ураженням екстрапірамідної системи, на тлі печінкової
- 97. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Диференціальний діагноз: Диференціальний діагноз. З гепатогенною енцефалопатією: хворіють люди різного віку, а
- 98. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Лікування: Лікування. До робіт Уемена та Камінгса (які пояснили біохімічний механізм цього
- 99. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Лікування: Позитивний ефект від лікування зазвичай досягається через 6-8 міс. Необхідно лікувати
- 100. ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ (ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА) Лікування, профілактика: Приклад лікування: 1) купреніл по 250 мг х б р.,
- 102. Скачать презентацию

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Група захворювань (якщо мова йде про первинну міопатію),
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Група захворювань (якщо мова йде про первинну міопатію),

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
З сучасних авторів багато працювали в цій галузі:
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
З сучасних авторів багато працювали в цій галузі:

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Класифікація міопатій:
(Л.О.Бадалян, 1973)
I. Первинні міопатії:
Юнацька (ювенільна) форма
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Класифікація міопатій:
(Л.О.Бадалян, 1973)
I. Первинні міопатії:
Юнацька (ювенільна) форма

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Класифікація міопатій:
(Л.О.Бадалян, 1973)
II. Вторинні міопатії (аміотрофії):
Спінальна аміотрофія
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Класифікація міопатій:
(Л.О.Бадалян, 1973)
II. Вторинні міопатії (аміотрофії):
Спінальна аміотрофія

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Класифікація міопатій
(Л.О.Бадалян, 1973)
IV. Міопатичні синдроми (фенокопії ПМД) при
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Класифікація міопатій
(Л.О.Бадалян, 1973)
IV. Міопатичні синдроми (фенокопії ПМД) при

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Не всі визнають розподіл м'язових дистрофій на
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Не всі визнають розподіл м'язових дистрофій на

ПЕРВИННІ МІОПАТІЇ:
Патологічна анатомія. При аутопсії: м'язи стоншені, жовто-сірого кольору в зв'язку
ПЕРВИННІ МІОПАТІЇ:
Патологічна анатомія. При аутопсії: м'язи стоншені, жовто-сірого кольору в зв'язку

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Етіологія і патогенез. Міопатії відносяться до групи спадкових
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Етіологія і патогенез. Міопатії відносяться до групи спадкових

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Етіологія і патогенез.
Поряд з чіткими спадковими формами міопатій
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Етіологія і патогенез.
Поряд з чіткими спадковими формами міопатій

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Патогенез ПМД на даний момент повністю не з'ясований.
Неврогенна
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Патогенез ПМД на даний момент повністю не з'ясований.
Неврогенна

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Патогенез ПМД.
При ПМД Дрейфуса (рідкісна форма з контрактурами,
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Патогенез ПМД.
При ПМД Дрейфуса (рідкісна форма з контрактурами,
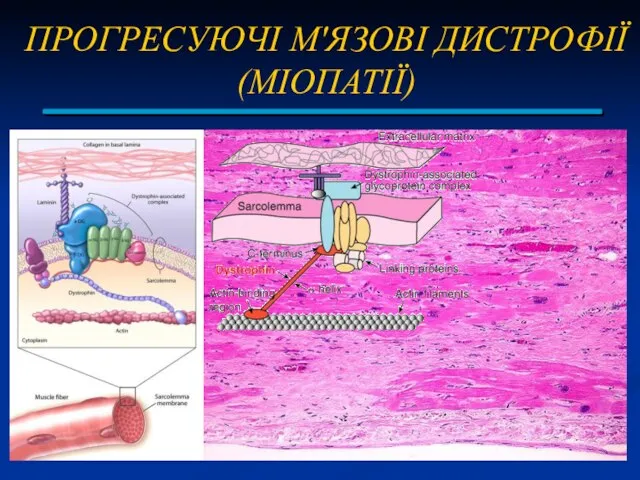
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Перебіг та симптоми. Головними клінічними симптомами міопатії є:
-повільно
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Перебіг та симптоми. Головними клінічними симптомами міопатії є:
-повільно
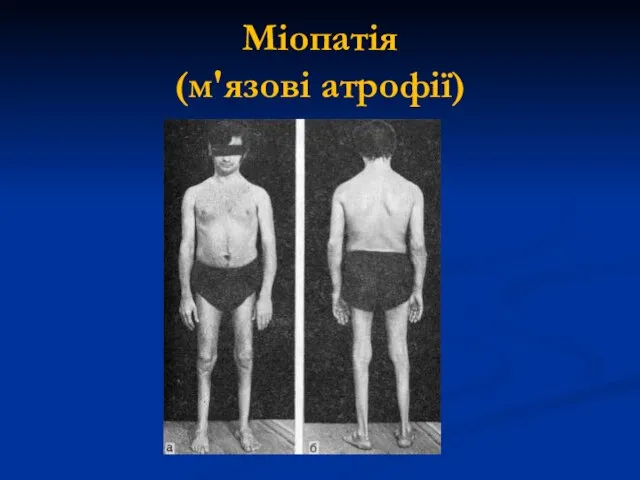
Міопатія
(м'язові атрофії)
Міопатія
(м'язові атрофії)
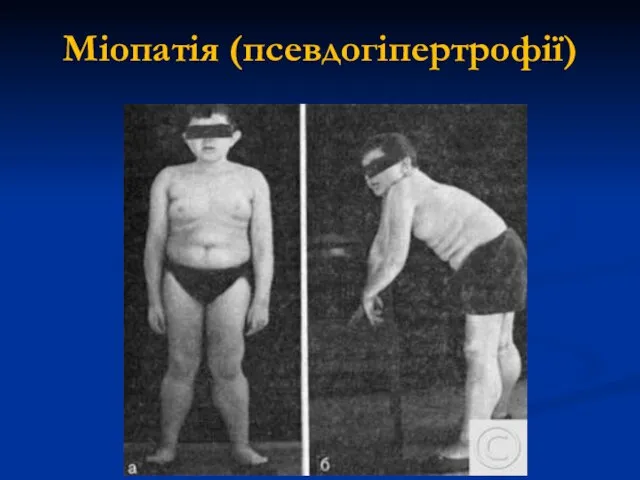
Міопатія (псевдогіпертрофії)
Міопатія (псевдогіпертрофії)

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Симптоми.
Атрофії розвиваються дуже повільно, вражаючи поступово (і зазвичай
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Симптоми.
Атрофії розвиваються дуже повільно, вражаючи поступово (і зазвичай

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Симптоми.
Характерно для міопатій: гладенький полірований лоб, недостатнє змикання
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Симптоми.
Характерно для міопатій: гладенький полірований лоб, недостатнє змикання
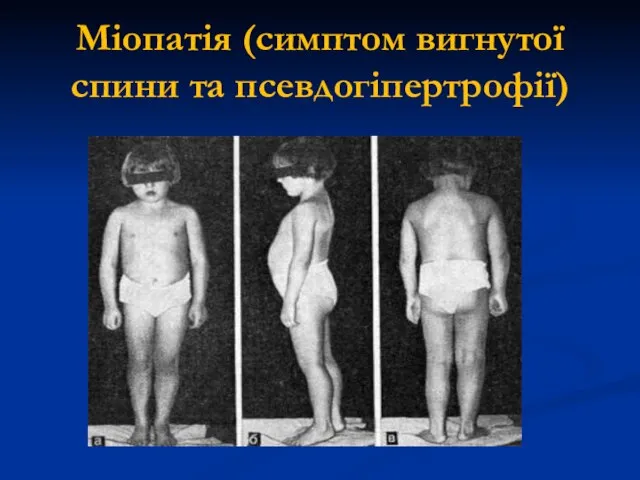
Міопатія (симптом вигнутої спини та псевдогіпертрофії)
Міопатія (симптом вигнутої спини та псевдогіпертрофії)
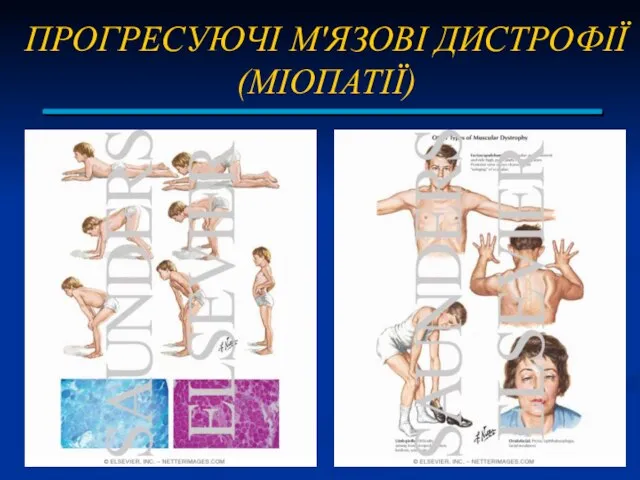
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
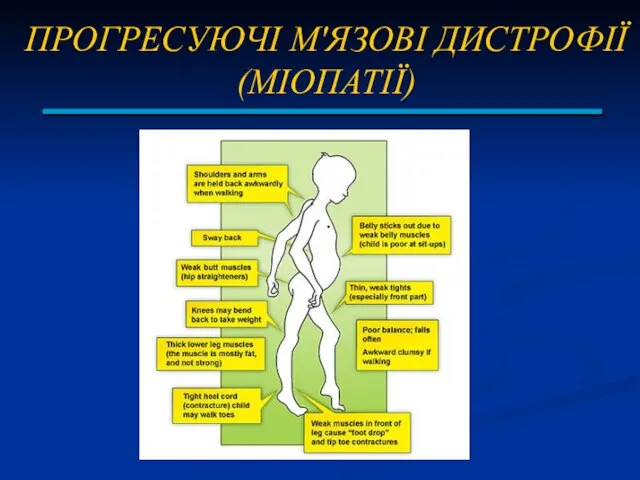
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)

Симптом «драбинки» - вставання драбинкою
Симптом «драбинки» - вставання драбинкою
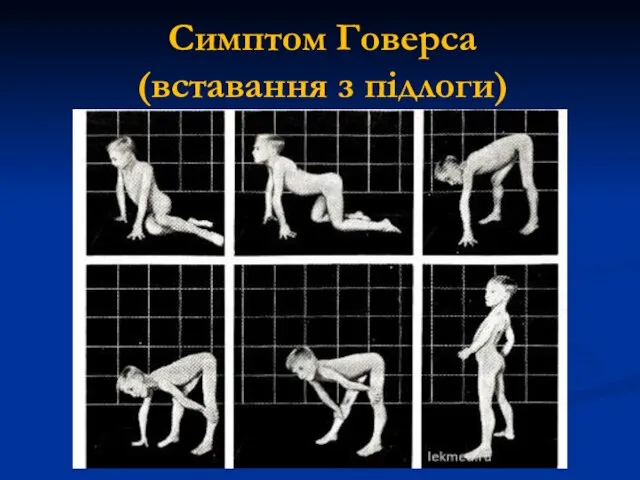
Симптом Говерса
(вставання з підлоги)
Симптом Говерса
(вставання з підлоги)

ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Симптоми.
Інтелект не страждає (якщо не брати до уваги
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
Симптоми.
Інтелект не страждає (якщо не брати до уваги
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)
ПРОГРЕСУЮЧІ М'ЯЗОВІ ДИСТРОФІЇ (МІОПАТІЇ)

ОКРЕМІ ФОРМИ МІОПАТІЙ
Ювенільна (юнацька) форма Ерба
це найбільш аморфна форма, більшість
ОКРЕМІ ФОРМИ МІОПАТІЙ
Ювенільна (юнацька) форма Ерба
це найбільш аморфна форма, більшість
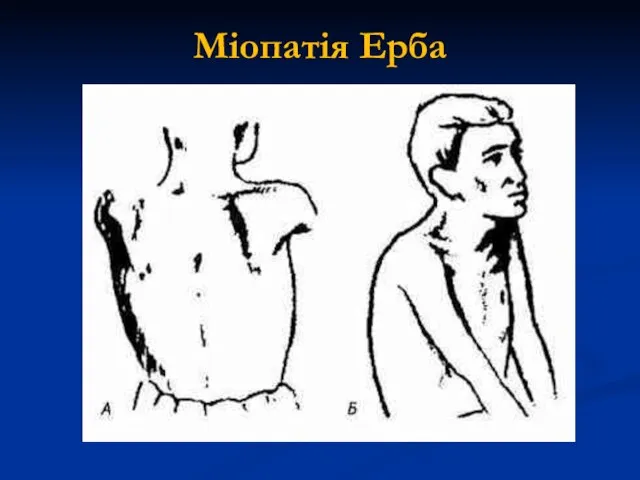
Міопатія Ерба
Міопатія Ерба

ОКРЕМІ ФОРМИ МІОПАТІЙ
Псевдогіпертрофічна форма Дюшена
хвороба успадковується за рецесивним типом, зчепленим
ОКРЕМІ ФОРМИ МІОПАТІЙ
Псевдогіпертрофічна форма Дюшена
хвороба успадковується за рецесивним типом, зчепленим
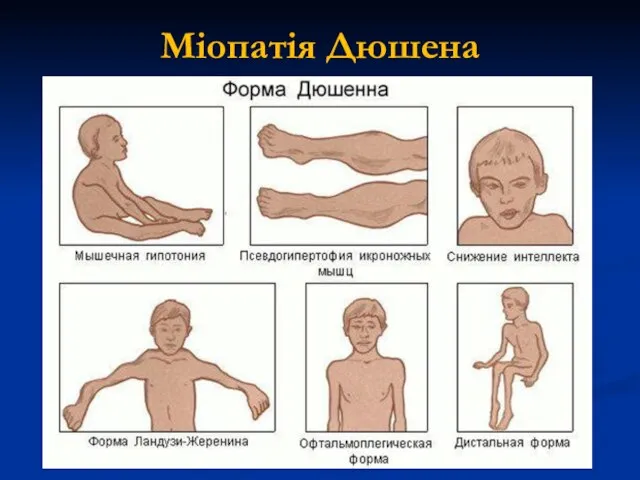
Міопатія Дюшена
Міопатія Дюшена
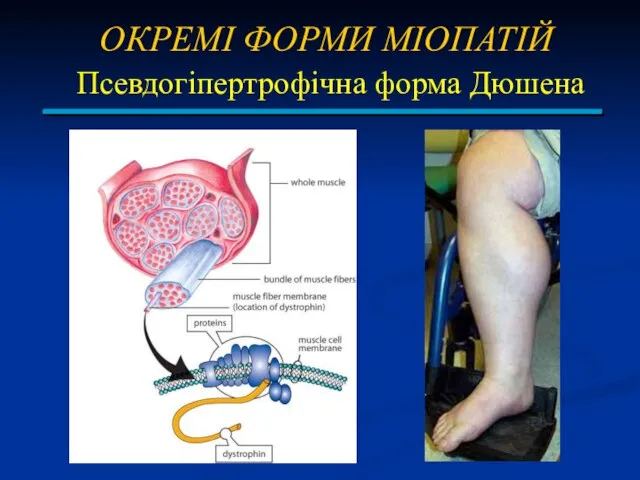
ОКРЕМІ ФОРМИ МІОПАТІЙ
Псевдогіпертрофічна форма Дюшена
ОКРЕМІ ФОРМИ МІОПАТІЙ
Псевдогіпертрофічна форма Дюшена
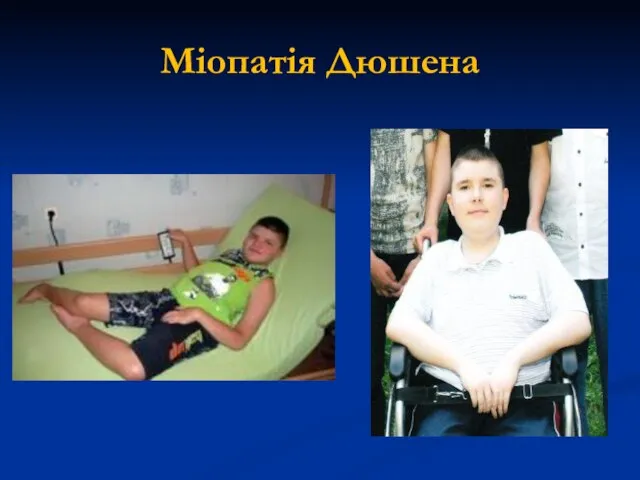
Міопатія Дюшена
Міопатія Дюшена

М'ЯЗОВА ДИСТРОФІЯ
БЕККЕРА-КІНЕРА
Х-зчеплена доброякісна міодистрофія, що була описана в 1955 році;
починається
М'ЯЗОВА ДИСТРОФІЯ
БЕККЕРА-КІНЕРА
Х-зчеплена доброякісна міодистрофія, що була описана в 1955 році;
починається
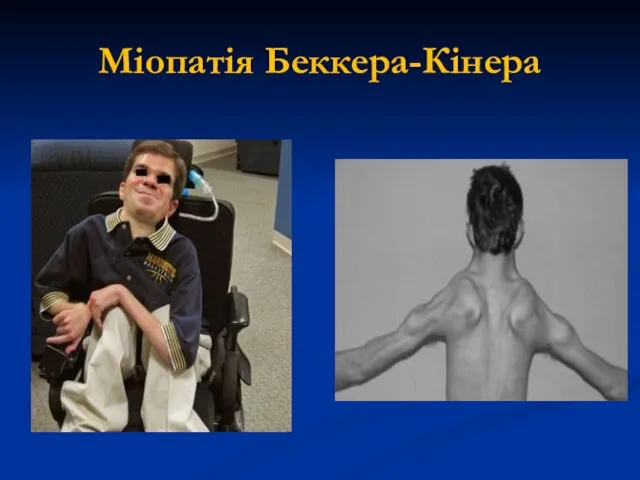
Міопатія Беккера-Кінера
Міопатія Беккера-Кінера

ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
Захворювання успадковується за аутосомно-домінантним типом з високою пенетрантністю і
ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
Захворювання успадковується за аутосомно-домінантним типом з високою пенетрантністю і
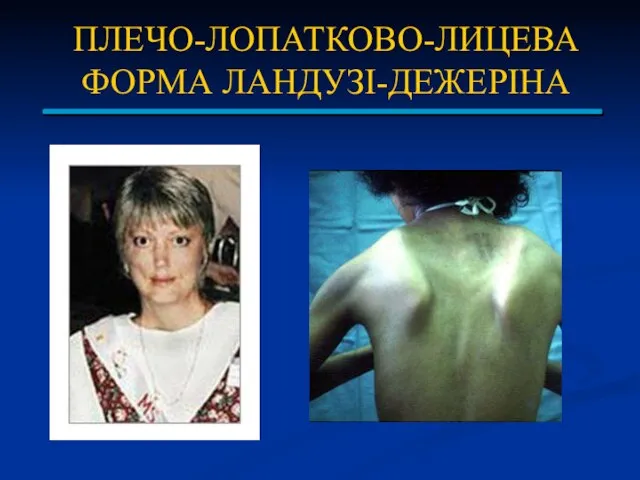
ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА

ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
Рідкісні форми:
- дистальна;
- окулярна;
- окулофарінгеальна.
ЕМГ вказує на ураження м'язів
ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
Рідкісні форми:
- дистальна;
- окулярна;
- окулофарінгеальна.
ЕМГ вказує на ураження м'язів
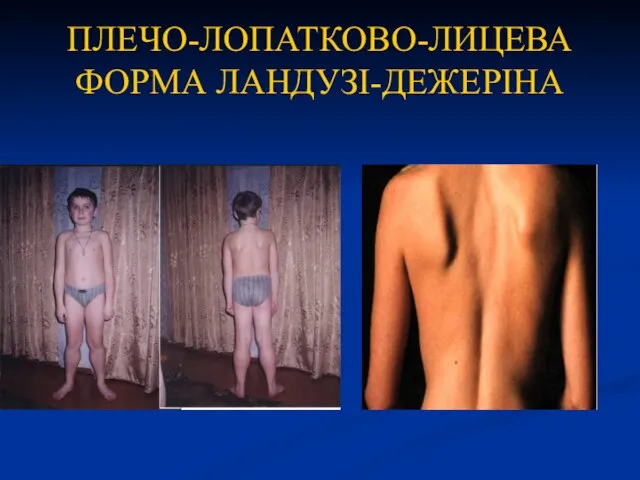
ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА
ПЛЕЧО-ЛОПАТКОВО-ЛИЦЕВА ФОРМА ЛАНДУЗІ-ДЕЖЕРІНА

Диференціальна діагностика міопатій
Іноді подібну клінічну картину має хвороба Вердніга-Гоффманна, але
Диференціальна діагностика міопатій
Іноді подібну клінічну картину має хвороба Вердніга-Гоффманна, але

ЛІКУВАННЯ МІОПАТІЙ
Лікування міопатії-повинно бути постійним або з короткими перервами.
Лікувати необхідно з
ЛІКУВАННЯ МІОПАТІЙ
Лікування міопатії-повинно бути постійним або з короткими перервами.
Лікувати необхідно з

ЛІКУВАННЯ МІОПАТІЙ
В той же час деякі автори не підтверджують позитивної дії
ЛІКУВАННЯ МІОПАТІЙ
В той же час деякі автори не підтверджують позитивної дії

ЛІКУВАННЯ МІОПАТІЙ
Зразок схеми курсів лікування ПМД за Бадаляновим Л.О.
I курс- АТФ
ЛІКУВАННЯ МІОПАТІЙ
Зразок схеми курсів лікування ПМД за Бадаляновим Л.О.
I курс- АТФ

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Описали Шарко і Марі в 1806 р.
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Описали Шарко і Марі в 1806 р.

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Найчастіше успадковується за домінантним типом зі значною
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Найчастіше успадковується за домінантним типом зі значною

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Патологічна анатомія.
Відзначається переродження осьових циліндрів периферичних нервів
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Патологічна анатомія.
Відзначається переродження осьових циліндрів периферичних нервів
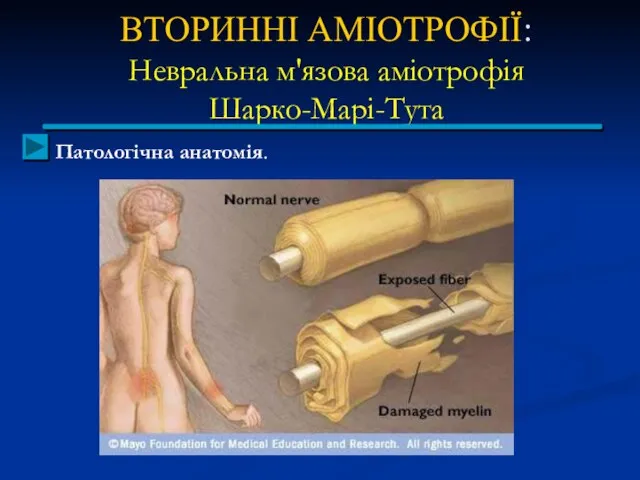
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Патологічна анатомія.
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Патологічна анатомія.

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка. Процес, перш за все, вражає стопи,
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка. Процес, перш за все, вражає стопи,
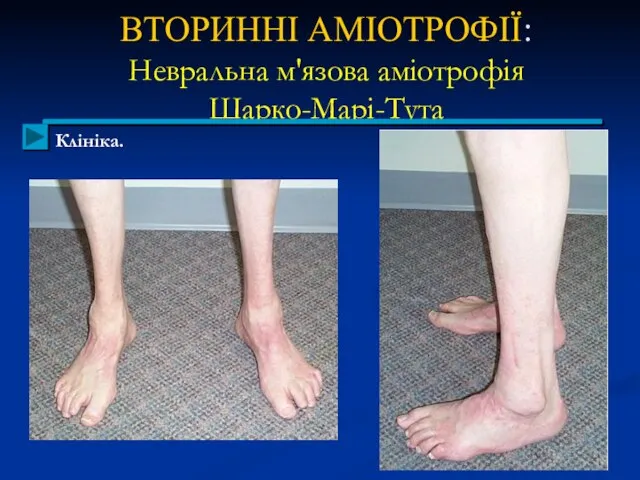
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка.
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка.

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка.
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка.
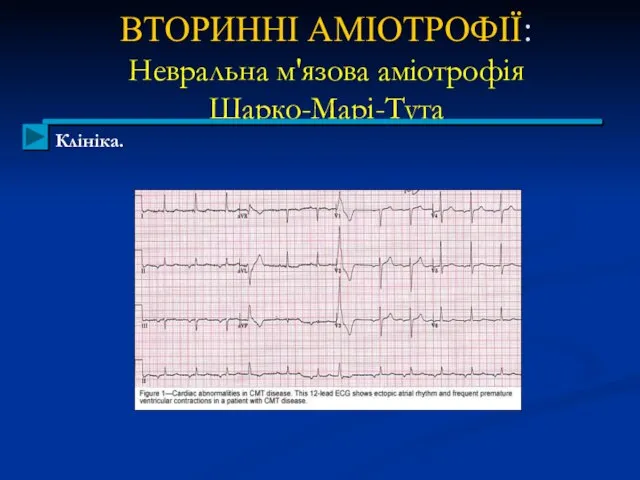
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка.
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка.

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка. Значно рідше першими в процес залучаються
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Клініка. Значно рідше першими в процес залучаються

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Перебіг захворювання повільний, але неухильний. Хворі довго
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Перебіг захворювання повільний, але неухильний. Хворі довго

ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Лікування:
вітаміни групи В;
АТФ;
антихолінестеразні препарати;
анаболічні стероїди;
Секуренін;
Аденіл в/м по
ВТОРИННІ АМІОТРОФІЇ:
Невральна м'язова аміотрофія
Шарко-Марі-Тута
Лікування:
вітаміни групи В;
АТФ;
антихолінестеразні препарати;
анаболічні стероїди;
Секуренін;
Аденіл в/м по

СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
Захворювання описав Вердніг в 1891р. Гоффман
СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
Захворювання описав Вердніг в 1891р. Гоффман

СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
При вродженій формі: майже відсутнє ворушіння
СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
При вродженій формі: майже відсутнє ворушіння
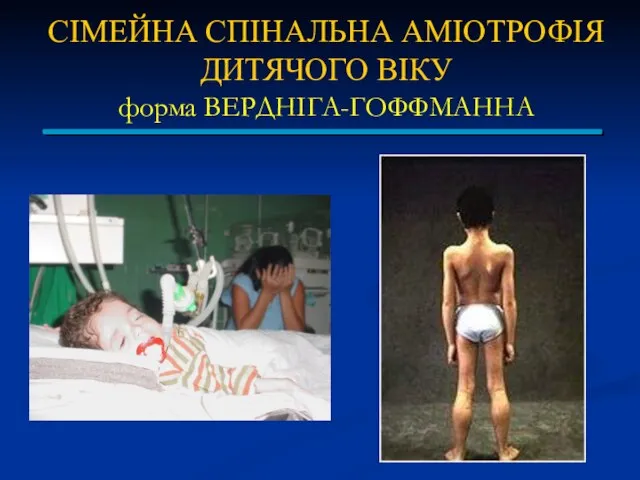
СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА

СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
Пізня форма. Зазвичай починається у дітей,
СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
Пізня форма. Зазвичай починається у дітей,

СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
Діагноз. Як правило хворі надходять з
СІМЕЙНА СПІНАЛЬНА АМІОТРОФІЯ ДИТЯЧОГО ВІКУ
форма ВЕРДНІГА-ГОФФМАННА
Діагноз. Як правило хворі надходять з

СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ (хвороба Арана- Дюшена)
Це дуже неясний розділ клінічної
СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ (хвороба Арана- Дюшена)
Це дуже неясний розділ клінічної

СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Клініка. Основний симптом захворювання - м'язова атрофія, що починається,
СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Клініка. Основний симптом захворювання - м'язова атрофія, що починається,

СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Таким чином, виділяють 3 форми спінальної аміотрофії:
хронічна дистальна;
хронічна проксимальна;
бульбарно-спінальна
СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Таким чином, виділяють 3 форми спінальної аміотрофії:
хронічна дистальна;
хронічна проксимальна;
бульбарно-спінальна

СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Діагноз. Звичайно, найбільші труднощі виникають при диференціальній діагностиці з
СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Діагноз. Звичайно, найбільші труднощі виникають при диференціальній діагностиці з

СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Лікування:
антихолінестеразні препарати,
стероїди,
вітаміни групи В та Е,
пірацетам,
аміналон,
піридітол,
електростимуляція.
СПІНАЛЬНІ АМІОТРОФІЇ
ДОРОСЛИХ
Лікування:
антихолінестеразні препарати,
стероїди,
вітаміни групи В та Е,
пірацетам,
аміналон,
піридітол,
електростимуляція.

МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Міотонія - особливий стан м'язів, що пояснюється в тому, що
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Міотонія - особливий стан м'язів, що пояснюється в тому, що

МІОТОНІЯ
ХВОРОБА ТОМСЕНА
За даними А.П.Зінченко з 110 хворих на міотонію тільки у
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
За даними А.П.Зінченко з 110 хворих на міотонію тільки у

МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Клініка. Хворий після стану спокою з великими труднощами починає рухатись,
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Клініка. Хворий після стану спокою з великими труднощами починає рухатись,

МІОТОНІЯ
ХВОРОБА ТОМСЕНА
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
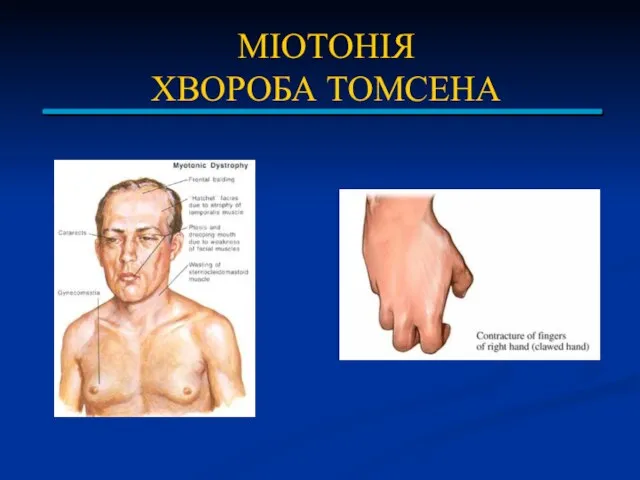
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
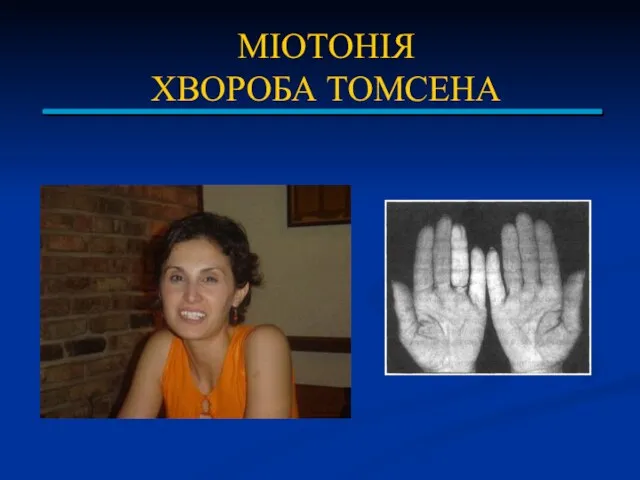
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
МІОТОНІЯ
ХВОРОБА ТОМСЕНА

МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Хворі відрізняються атлетичною статурою. М'язи їх на дотик щільні та
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Хворі відрізняються атлетичною статурою. М'язи їх на дотик щільні та

МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Лікування.
Застосовують теплі ванни та інші теплові процедури, помірну ЛФК.
Шінявський та
МІОТОНІЯ
ХВОРОБА ТОМСЕНА
Лікування.
Застосовують теплі ванни та інші теплові процедури, помірну ЛФК.
Шінявський та

Спадкові захворювання нервової системи
(хвороба Штрюмпелля, атаксії, хорея Гентінґтона, хвороба Вільсона-Коновалова)
ЗАПОРІЗЬКИЙ ДЕРЖАВНИЙ
Спадкові захворювання нервової системи
(хвороба Штрюмпелля, атаксії, хорея Гентінґтона, хвороба Вільсона-Коновалова)
ЗАПОРІЗЬКИЙ ДЕРЖАВНИЙ

СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Пат. анатомія, патогенез
Хронічно прогресуюче спадкове захворювання. Вперше описав
СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Пат. анатомія, патогенез
Хронічно прогресуюче спадкове захворювання. Вперше описав

СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Клінічна картина:
Клініка. Частіше хворіють на 2-му десятиріччі
СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Клінічна картина:
Клініка. Частіше хворіють на 2-му десятиріччі

СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Клінічна картина:
Так, за даними Ozvath з 267
СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Клінічна картина:
Так, за даними Ozvath з 267

СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Діагноз
У типових випадках за наявністю сімейного анамнезу
СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Діагноз
У типових випадках за наявністю сімейного анамнезу

СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Лікування:
Зниження м'язового тонусу:
ліорезал в ін'єкціях (Швейцарія),
баклофен 1таблетка
СІМЕЙНА СПАСТИЧНА ПАРАПЛЕГІЯ ШТРЮМПЕЛЛЯ
Лікування:
Зниження м'язового тонусу:
ліорезал в ін'єкціях (Швейцарія),
баклофен 1таблетка

СПАДКОВІ АТАКСІЇ
Спадкові атаксії зустрічаються в різних формах і в кожній формі
СПАДКОВІ АТАКСІЇ
Спадкові атаксії зустрічаються в різних формах і в кожній формі

СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Пат. анатомія, патогенез:
Описана цим автором в 1861 р.
СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Пат. анатомія, патогенез:
Описана цим автором в 1861 р.

СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Клінічна картина:
Характерні екстраневральні порушення:
А) зміна скелета: кіфосколіоз, стопи
СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Клінічна картина:
Характерні екстраневральні порушення:
А) зміна скелета: кіфосколіоз, стопи

СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Діагноз:
Діагноз в типових випадках не складний, але типові
СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Діагноз:
Діагноз в типових випадках не складний, але типові

СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Лікування:
Лікування симптоматичне:
- Френелевська гімнастика,
- Вітаміни групи В,
- Препарати
СІМЕЙНА
АТАКСІЯ ФРІДРАЙХА
Лікування:
Лікування симптоматичне:
- Френелевська гімнастика,
- Вітаміни групи В,
- Препарати

МОЗОЧКОВА МІОКЛОНІЧНА ДИССИНЕРГІЯ ХАНТА
Пат. анатомія, клініка, лікування.
Описана Хантом в 1921р.
Пат
МОЗОЧКОВА МІОКЛОНІЧНА ДИССИНЕРГІЯ ХАНТА
Пат. анатомія, клініка, лікування.
Описана Хантом в 1921р.
Пат

ХОРЕЯ ГЕНТІНҐТОНА
(ХОРЕІЧНА ДЕМЕНЦІЯ)
Пат. анатомія, патогенез:
Хронічне прогресуюче захворювання, основними ознаками якого
ХОРЕЯ ГЕНТІНҐТОНА
(ХОРЕІЧНА ДЕМЕНЦІЯ)
Пат. анатомія, патогенез:
Хронічне прогресуюче захворювання, основними ознаками якого

ХОРЕЯ ГЕНТІНҐТОНА
Клініка:
Клінічна картина. Гіперкінез проявляється швидкими неритмічними довільними рухами, що
ХОРЕЯ ГЕНТІНҐТОНА
Клініка:
Клінічна картина. Гіперкінез проявляється швидкими неритмічними довільними рухами, що

ХОРЕЯ ГЕНТІНҐТОНА
Клініка:
Приблизно у 10-15% хворих на ХГ є акінетико-ригідна форма.
ХОРЕЯ ГЕНТІНҐТОНА
Клініка:
Приблизно у 10-15% хворих на ХГ є акінетико-ригідна форма.

ХОРЕЯ ГЕНТІНҐТОНА
Клініка:
Клініка. Основний симптом - це локомоторна та статична атаксія.
ХОРЕЯ ГЕНТІНҐТОНА
Клініка:
Клініка. Основний симптом - це локомоторна та статична атаксія.

ХОРЕЯ ГЕНТІНҐТОНА
Діагноз:
Діагноз і диференційний діагноз.
Поєднання хореїформного гіперкінезу з деменцією, пізній
ХОРЕЯ ГЕНТІНҐТОНА
Діагноз:
Діагноз і диференційний діагноз.
Поєднання хореїформного гіперкінезу з деменцією, пізній

ХОРЕЯ ГЕНТІНҐТОНА
Лікування:
Необхідно: блокувати дофамінергічні рецептори або зменшити вміст дофаміну (у
ХОРЕЯ ГЕНТІНҐТОНА
Лікування:
Необхідно: блокувати дофамінергічні рецептори або зменшити вміст дофаміну (у

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Пат. анатомія, патогенез:
У 1883 р. Штромпелль описав захворювання, яке
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Пат. анатомія, патогенез:
У 1883 р. Штромпелль описав захворювання, яке

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Клініка:
Клінічна картина. Неврологічні симптоми розвиваються поступово: спочатку порушується м'язовий
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Клініка:
Клінічна картина. Неврологічні симптоми розвиваються поступово: спочатку порушується м'язовий

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Клініка:
психічні розлади при цій хворобі вельми різноманітні. Хворі
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Клініка:
психічні розлади при цій хворобі вельми різноманітні. Хворі
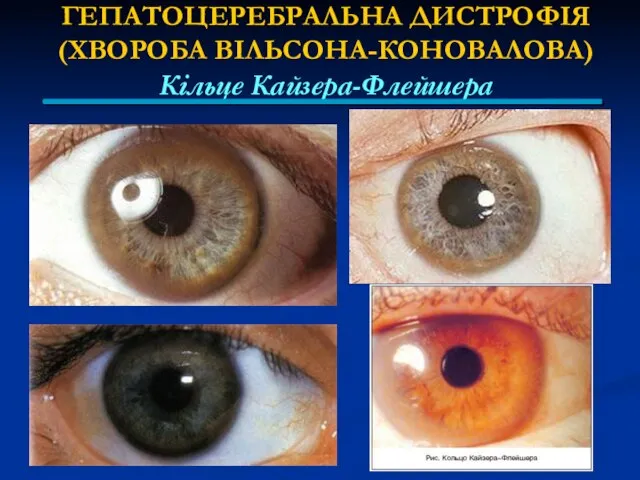
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Кільце Кайзера-Флейшера
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Кільце Кайзера-Флейшера

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Етіологія, пат. анатомія, патогенез, клініка:
Крім мозкових явищ важливе місце
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Етіологія, пат. анатомія, патогенез, клініка:
Крім мозкових явищ важливе місце

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Єтіологія, пат. анатомія, патогенез, клініка:
Вочевидь батько і мати є
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Єтіологія, пат. анатомія, патогенез, клініка:
Вочевидь батько і мати є

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Що ж власне передається у спадок?
Який патогенез хвороби?
При цій
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Що ж власне передається у спадок?
Який патогенез хвороби?
При цій

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Діагноз:
Діагноз. Прогресуюче захворювання, з переважним ураженням екстрапірамідної системи, на
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Діагноз:
Діагноз. Прогресуюче захворювання, з переважним ураженням екстрапірамідної системи, на

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Диференціальний діагноз:
Диференціальний діагноз.
З гепатогенною енцефалопатією: хворіють люди різного
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Диференціальний діагноз:
Диференціальний діагноз.
З гепатогенною енцефалопатією: хворіють люди різного

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Лікування:
Лікування. До робіт Уемена та Камінгса (які пояснили біохімічний
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Лікування:
Лікування. До робіт Уемена та Камінгса (які пояснили біохімічний

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Лікування:
Позитивний ефект від лікування зазвичай досягається через 6-8 міс.
Необхідно
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Лікування:
Позитивний ефект від лікування зазвичай досягається через 6-8 міс.
Необхідно

ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Лікування, профілактика:
Приклад лікування:
1) купреніл по 250 мг х б
ГЕПАТОЦЕРЕБРАЛЬНА ДИСТРОФІЯ
(ХВОРОБА ВІЛЬСОНА-КОНОВАЛОВА)
Лікування, профілактика:
Приклад лікування:
1) купреніл по 250 мг х б
 Дәрігер-науқас-мейірбеке қарым-қатнасы
Дәрігер-науқас-мейірбеке қарым-қатнасы Диабетическая ретинопатия
Диабетическая ретинопатия Обзор клинического случая
Обзор клинического случая Острая сердечно-сосудистая недостаточность
Острая сердечно-сосудистая недостаточность Влияние образа жизни на индивидуальное и общественное здоровье. Семья и формирование ЗОЖ
Влияние образа жизни на индивидуальное и общественное здоровье. Семья и формирование ЗОЖ Функціональна анатомія серця
Функціональна анатомія серця Функциональная анатомия эндокринных органов
Функциональная анатомия эндокринных органов Понятие и роль биостатистики как основной составляющей системы доказательной медицины
Понятие и роль биостатистики как основной составляющей системы доказательной медицины Рабочая концепция одаренности
Рабочая концепция одаренности Питание женщины во время беременности
Питание женщины во время беременности Уход за тяжелыми больными
Уход за тяжелыми больными Наркомания и подросток
Наркомания и подросток Репродуктивное здоровье. Таблица
Репродуктивное здоровье. Таблица Медициналық қызмет сапасына ішкі және сыртқы сараптау жүргізу және ұйымдастыру ережелері
Медициналық қызмет сапасына ішкі және сыртқы сараптау жүргізу және ұйымдастыру ережелері Первая помощь при укусах животных
Первая помощь при укусах животных Кодификаторы и классификаторы в здравоохранении. МКБ-10. Учет и отчетность заболеваемости в Российской Федерации
Кодификаторы и классификаторы в здравоохранении. МКБ-10. Учет и отчетность заболеваемости в Российской Федерации Острые заболевания наружного уха
Острые заболевания наружного уха Правила организации хранения товаров аптечного ассортимента. Требования к помещениям хранения товаров аптечного ассортимента
Правила организации хранения товаров аптечного ассортимента. Требования к помещениям хранения товаров аптечного ассортимента Правила личной гигиены
Правила личной гигиены Щитовидная железа
Щитовидная железа Диференціальна діагностика захворювань із синдромом екзантеми
Диференціальна діагностика захворювань із синдромом екзантеми Теории детской и возрастной психологии
Теории детской и возрастной психологии Болезнь Помпе и Нимана-Пика
Болезнь Помпе и Нимана-Пика Физиологические механизмы трудовой деятельности и приспособления организма к изменившимся условиям. Лекция 39
Физиологические механизмы трудовой деятельности и приспособления организма к изменившимся условиям. Лекция 39 Геморрагический синдром. Острый лейкоз
Геморрагический синдром. Острый лейкоз Синдром слабости синусового узла. Синусовая брадикардия
Синдром слабости синусового узла. Синусовая брадикардия Педиатрия, учение о детских болезнях
Педиатрия, учение о детских болезнях Алгоритм работы при отработке заболевших Сovid-19
Алгоритм работы при отработке заболевших Сovid-19