- Наследственные Нарушение(педиатрия)

Содержание
- 2. НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА ВЕЩЕСТВ К настоящему времени известно более 2500 наследственных ферментопатий, 20 классов наследственных болезней
- 3. Клинические проявления наследственных болезней обмена веществ во многом определяются поражением нервной системы (особенно при нарушениях обмена
- 4. Наследственные болезни обмена аминокислот Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты являются основными структурными элементами
- 5. Для обеспечения нормального роста и развития важно не только количество поступающих аминокислот, но и их соотношение.
- 6. Наследственные нарушения обмена аминокислот (Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.) 1. Наследственные нарушения обмена аминокислот, сопровождающиеся
- 7. 3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и др. К этой группе относятся
- 8. Фенилкетонурия (ФКУ) Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность". Тип наследования - аутосомнорецессивный.
- 9. Патофизиология Избыточно потребляемый фенилаланин (т.е. не использующийся для синтеза белка), как правило, преобразуется в тирозин фенилаланингидроксилазу;
- 10. Варианты заболевания Хотя почти все случаи (98–99%) ФКУ развиваются в результате дефицита фенилаланингидроксилазы, фенилаланин также может
- 11. Клинические проявления Большинство детей с фенилкетонурией при рождении нормальные, симптомы и признаки появляются постепенно, в течение
- 12. Диагностика Рутинный неонатальный скрининг Уровни фенилаланина В США и многих развитых странах, всех новорожденных обследуют на
- 13. Прогноз Адекватное лечение, начатое в первые дни жизни, предотвращает возникновение тяжелых проявлений заболевания. Тем не менее
- 14. Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы. Различают следующие формы
- 15. Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная
- 16. Все заболевания в зависимости от нарушения вида обмена веществ можно разделить на следующие группы: нарушения обмена
- 17. По типу наследования выделяют следующие варианты: Моногенные болезни с аутосомно-доминантным типом наследования (болезнь Марфана) Моногенные болезни
- 19. Скачать презентацию

НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ
ОБМЕНА ВЕЩЕСТВ
К настоящему времени известно более 2500 наследственных ферментопатий,
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ
ОБМЕНА ВЕЩЕСТВ
К настоящему времени известно более 2500 наследственных ферментопатий,

Клинические проявления наследственных болезней обмена веществ во многом определяются поражением нервной

Наследственные болезни
обмена аминокислот
Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты
Наследственные болезни
обмена аминокислот
Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты

Для обеспечения нормального роста и развития важно не только количество поступающих
Для обеспечения нормального роста и развития важно не только количество поступающих

Наследственные нарушения обмена аминокислот
(Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.)
1. Наследственные нарушения
Наследственные нарушения обмена аминокислот
(Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.)
1. Наследственные нарушения

3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и
3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и

Фенилкетонурия (ФКУ)
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность".
Фенилкетонурия (ФКУ)
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность".

Патофизиология
Избыточно потребляемый фенилаланин (т.е. не использующийся для синтеза белка), как правило,
Патофизиология
Избыточно потребляемый фенилаланин (т.е. не использующийся для синтеза белка), как правило,

Варианты заболевания
Хотя почти все случаи (98–99%) ФКУ развиваются в результате дефицита
Варианты заболевания
Хотя почти все случаи (98–99%) ФКУ развиваются в результате дефицита

Клинические проявления
Большинство детей с фенилкетонурией при рождении нормальные, симптомы и признаки
Клинические проявления
Большинство детей с фенилкетонурией при рождении нормальные, симптомы и признаки

Диагностика
Рутинный неонатальный скрининг
Уровни фенилаланина
В США и многих развитых странах, всех новорожденных
Диагностика
Рутинный неонатальный скрининг
Уровни фенилаланина
В США и многих развитых странах, всех новорожденных

Прогноз
Адекватное лечение, начатое в первые дни жизни, предотвращает возникновение тяжелых проявлений
Прогноз
Адекватное лечение, начатое в первые дни жизни, предотвращает возникновение тяжелых проявлений

Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью

Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы

Все заболевания в зависимости от нарушения вида обмена веществ можно разделить
Все заболевания в зависимости от нарушения вида обмена веществ можно разделить

По типу наследования выделяют следующие варианты:
Моногенные болезни с аутосомно-доминантным типом наследования
По типу наследования выделяют следующие варианты:
Моногенные болезни с аутосомно-доминантным типом наследования
 Неочікувана радість
Неочікувана радість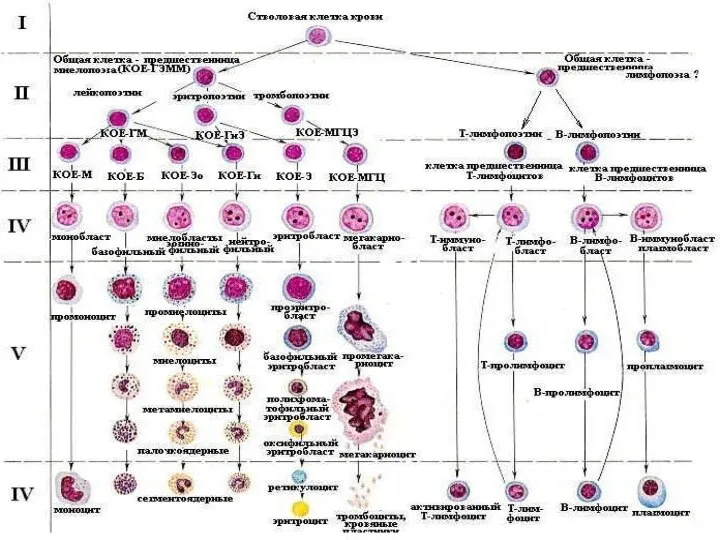 Болезни системы крови
Болезни системы крови ВНЧС. Заболевания височно-нижнечелюстного сустава
ВНЧС. Заболевания височно-нижнечелюстного сустава Свойства вакцины Oxford-AstraZeneca (AZD1222)
Свойства вакцины Oxford-AstraZeneca (AZD1222)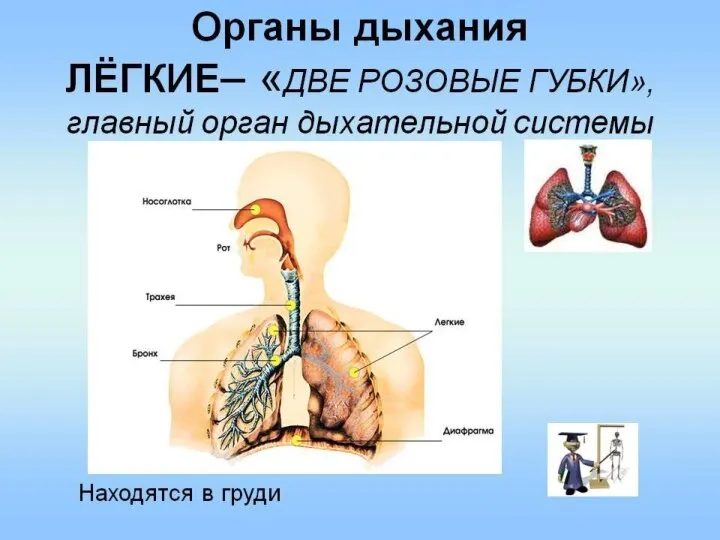 Органы дыхания
Органы дыхания Болезни органов дыхания
Болезни органов дыхания Ветеринарная клиника
Ветеринарная клиника Энцефалиты
Энцефалиты Жатырдан тыс жүктілікке жасалған хирургиялық емнен кейінгі науқастардың реабилитациясы
Жатырдан тыс жүктілікке жасалған хирургиялық емнен кейінгі науқастардың реабилитациясы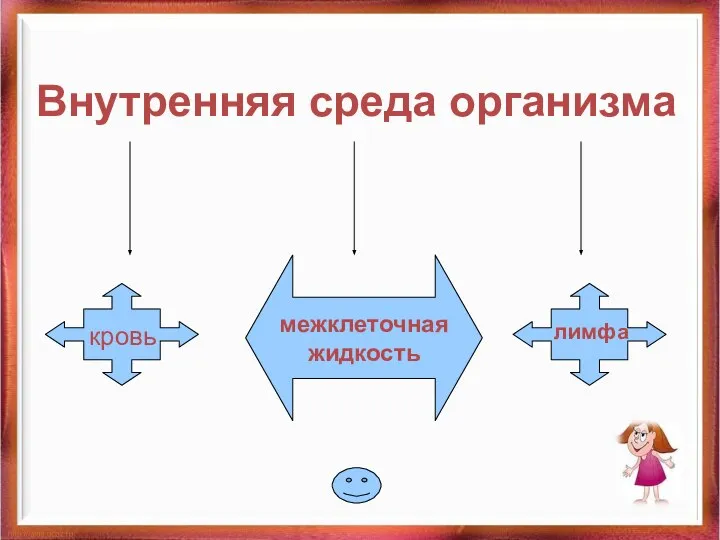 Внутренняя среда организма
Внутренняя среда организма Диффузные заболевания соединительной ткани. Дерматологические аспекты
Диффузные заболевания соединительной ткани. Дерматологические аспекты Физико-химические свойства лекарственных препаратов железа
Физико-химические свойства лекарственных препаратов железа Фармацевтическая биоэтика и биобезопасность
Фармацевтическая биоэтика и биобезопасность Беседа о гипер и гипогликемических состояниях
Беседа о гипер и гипогликемических состояниях Covid-19. Выплаты стимулирующего характера
Covid-19. Выплаты стимулирующего характера Типы пациента в реабилитации зависимости
Типы пациента в реабилитации зависимости Современное представление о патогенезе болезни Альцгеймера
Современное представление о патогенезе болезни Альцгеймера Диагностика типов привязанности
Диагностика типов привязанности Эрозивный гастрит. Клинический случай
Эрозивный гастрит. Клинический случай Гигиена кожных покровов
Гигиена кожных покровов Туберкулез полости рта. (Лекция 5)
Туберкулез полости рта. (Лекция 5) Психология и педагогика высшей школы
Психология и педагогика высшей школы Регионарная анестезия. Спинномозговая. Эпидуральная
Регионарная анестезия. Спинномозговая. Эпидуральная Bronchitis
Bronchitis Технологические особенности приготовления блюд из капусты для организации питания лиц страдающих заболеванием сердца и сосудов
Технологические особенности приготовления блюд из капусты для организации питания лиц страдающих заболеванием сердца и сосудов Вегетативна нервова система. Синдроми ураження різних відділів вегетативної нервової системи
Вегетативна нервова система. Синдроми ураження різних відділів вегетативної нервової системи Ультразвуковая допплерография в стоматологии
Ультразвуковая допплерография в стоматологии Практические аспекты применения ингибиторов PCSK-9 у пациентов высокого и очень высокого сердечно-сосудистого риска
Практические аспекты применения ингибиторов PCSK-9 у пациентов высокого и очень высокого сердечно-сосудистого риска